COMPADRE (COmparison of Multiple Protein sequence Alignments using Database RElationships) is a tool to detect protein sequence homologs. It can assess the relationship between query and database template by considering the similarity between query and template's known homologs.
Overview of homology detection procedure
COMPADRE is a method that can detect protein sequence homologs by considering the similarity between query and template's known homologs. COMPADRE will return a homolog ranking list for the input query sequence and the local sequence alignments for the query sequence and top templates. Firstly, the input sequence is fed into PROCAIN [1] (our previously reported method for sensitive detection of sequence homology). Secondly, COMPADRE modifies PROCAIN scores for the similarity between query and template by considering the template's homologs within the database. A linear combination of the original score for the given template with the scores for a broader protein family defined by structure, function, and sequence relationships to the template are used. Finally, a homolog ranking list and top homolog local alignments will be shown in the result webpage.
Currently, template database is composed of filted 5116 domains from SCOP representatives in ASTRAL 1.75 version(<20% identity)[2].
COMPADRE input
A single sequence or alignment of muliple protein sequences can be input to COMPADRE.
Note: It takes longer if seuqnece with long length or many homologs.
FASTA; or simple single line.
Example:
MASVSELACIYSALILHDDEVTVTEDKINALIKAAGVNVEPFWPGLFAKALANVNIGSLICNVGAGGPA
(2) Possible formats for alignments: (Don't forget to select "multiple sequence alignment" above the input frame!)
FASTA, ClustalW, Stockholm, or "simple" format :
name start(optional) sequence_segment end(optional)
name start(optional) sequence_segment end(optional)
...
Example (see complete alignments as input, click here.)
Note:
1. Single-letter notation for 20 amino acid types and two symbols for gaps (- or .) is used. Special letters of amino acid alphabet (BJUZXbjuzx) are treated as alanine in the search. Non-standard symbols are treated as gaps.
2. Alignments in all rows must be padded with dashes (-) to make them equal lengths.
3. By default, this PSI-BLAST search is performed only if query is a single sequence. If multiple sequence alignment is submitted, COMPADRE will use it 'as is', unless the user specifically requires the initial PSI-BLAST search.
Input Email: COMPADRE jobs usually take minutes to several hours for homolog setection. It takes longer if seuqnece with long length or many homologs. Thus, it is highly recommended to provide an email address so that the link to your result can be sent to you when the alignment is finished.
Input job name: Assign your sequences a short name can help identify your alignment job. This name will appear in the subject line of the email sent to you. Note: Job ID can only contain number, letter, underscore(_) and hyphen(-).
COMPADRE output
COMPADRE web server provides results including the following information.
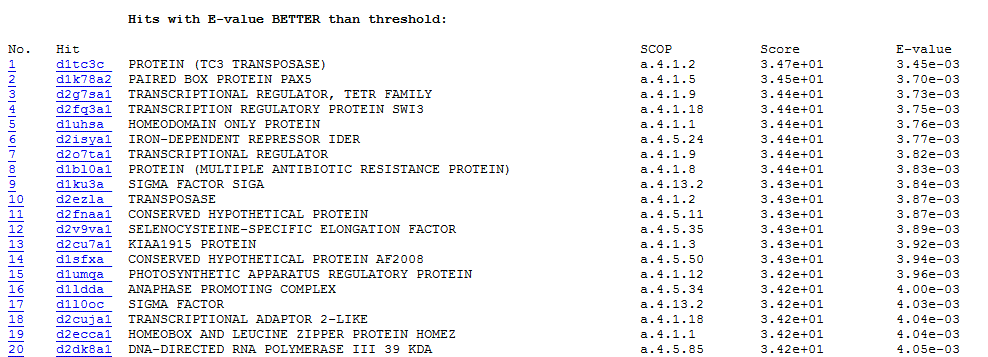
1. A homolog ranking list based on similarity.
Top hit is the closest homolog of the query sequence. The entire ranking list is divided into two parts: "Hits with E-value BETTER than threshold" and "Hits with E-value WORSE than threshold". The default e-value threshold is 0.005 and the default ranking list number is 100 (maximum is 5116). Users can parameterize them.
For each homolog, clickable ranking No. links to local alignments between query and template. Template domain name and SCOP classification are provided. The clickable domain name links to its SCOP webpage. The final similarity scores and e-values scores are also listed.
Output Ranking List Example (part)
2. Detailed local alignment between query and each homolog.
Below the entire list, the details of template protein name/explanation and the local alignment are shown. In order to show long local alignment, PROMALS alignment is provided. There is a button below linking to the original PROCAIN local alignment. The template structures in Jmol are provided for the top 10 Hits with E-value better than threshold.
For the local alignment, the first row and the last row marked as "SS_pred" represent the PSI-PRED [3] secondary structure prediction for the query and the template, respectively. The rows marked as "CONSENSUS" (linking to the entire profile) show the highest frequent residues of each position in the profile . "+" means the aligned position has a positive sequence profiling score, which suggests the possible conserved positions.
One Homolog Local Alignment Example (No.1 in the ranking list)
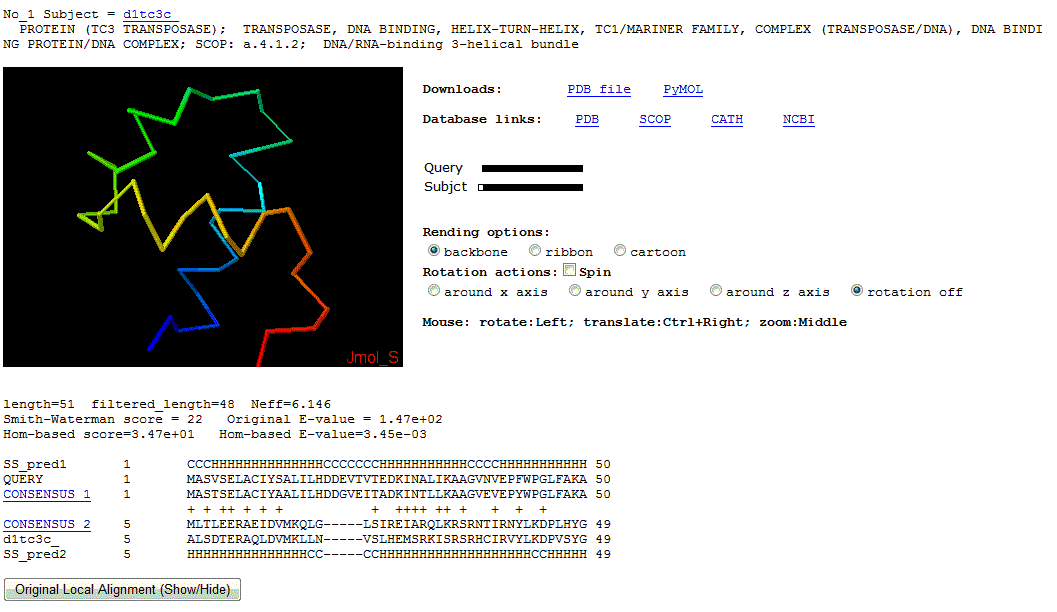
See one complete output example, click here.
Parameters are listed below
1. Input searching options.
Run PSI-BLAST if input is a sequence/an alignment
Run PSI-BLAST on query
To broaden the set of sequences representing the query family, the input MSA (or single sequence) can be used to initiate PSI-BLAST search [4]. The sequences detected by PSI-BLAST are filtered by user-specified E-value, coverage of the query length, and sequence identity to the query. The resulting PSI-BLAST alignment is submitted to COMPADRE as a query.
Note:
By default, this PSI-BLAST search is performed only if query is a single sequence. If multiple sequence alignment is submitted, COMPADRE will use it 'as is', unless the user specifically requires the initial PSI-BLAST search.
Run PSI-BLAST if input is a sequence
Prior to the COMPADRE search, runs PSI-BLAST to generate an alignment of homologs
if the submitted query is a single sequence. The resulting alignment is then used
as a query for COMPADRE.
Default = Yes
Run PSI-BLAST if input is an alignment
Prior to the COMPADRE search, runs PSI-BLAST to generate potentially more representative alignment of homologs if the submitted query is a multiple sequence alignment. Starting with the submitted alignment as a query, PSI-BLAST generates a new alignment of detected sequence homologs. This new alignment is then used as a query for the COMPADRE search.
Default = No
PSI-BLAST Iterations
Maximal number of PSI-BLAST iterations performed on query. Default = 5
PSI-BLAST Evalue
Maximal E-value for sequence homologs to be included in the PSI-BLAST alignment used as a query for the COMPADRE search. Default = 0.001
PSI-BLAST Hit Coverage
Minimal percentage of the query length covered by a PSI-BLAST hit required for the hit to be included in the PSI-BLAST alignment that is used for the COMPADRE search. Default = 20
Sequence identity of PSI-BLAST hit
Minimal percent sequence identity between a PSI-BLAST hit and the query required fot the hit to be included in the alignment that is used for the COMPADRE search. Default = 0
PSI-BLAST composition-based statistics
In PSI-BLAST, amino acid substitution matrices may be adjusted to compensate for biases in amino acid composition of the compared sequences. "Composition-based statistics" is the procedure of scaling all substitution scores by an analytically determined constant, while leaving the gap scores fixed. Default = YES
PSI-BLAST low complexity filtering
Masks off segments of the PSI-BLAST query that have low compositional complexity (e.g., common acidic-, basic- or proline-rich regions) detected by the SEG method. Default = NO
Gap fraction threshold (0.0 to 1.0)
Upper threshold of effective gap content in alignment columns used in the construction of profile-profile alignment. If a column contains too many gaps, it is disregarded in the process of profile comparison. Such columns are shown in the output as lower-case letters (residues) and dots (gaps). Default = 0.5
Gap opening penalty (Integer)
Score penalty for opening a new gap during the construction of profile-profile alignment. Default = 10
Gap extension penalty (Integer)
Score penalty for the extension of existing gap during the construction of profile-profile alignment. Default = 1
2. Output formatting options.
Expect Evalue
Maximal threshold of E-value for a hit to be displayed. Default = 0.1
Significance Evalue Threshold
Maximal E-value threshold for a hit to be considered reliable. Default = 0.005
Display up to (Integer)
Maximal number of hits (sorted by E-value) to be displayed. Maximum is the 5116 (database size). Default = 100
Width of alignment segment
Allows showing a long profile-profile alignment as multiple shorter segments, each containing a specified number of aligned positions. Default = 60
References
1. Yong W., Ruslan I.S. and Nick N.G. PROCAIN: protein profile comparison with assisting information. (2009) Nucleic Acids Res, 37(11), 3522-30.
2. Murzin A.G., Brenner S.E., Hubbard T., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. (1995) J Mol Biol 247(4),536-540.
3. Jones D.T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol , 292:195-202.
4. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. (2005) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389-3402.
If you feel helpful, please cite:
Jing Tong, Jimin Pei, Ruslan I. Sadreyev and Nick V. Grishin, "Known relations within search database enhance protein homology detection", 2014, In Preparation
Comments, suggestions and bug reports to: