d1gc5a_d1omha_
Motif
(click icon to enlarge) | SCOPid | Range | SCOP Class | SCOP Fold | SCOP Superfamily | SCOP Family | Protein |
|---|
1gc5_Ah
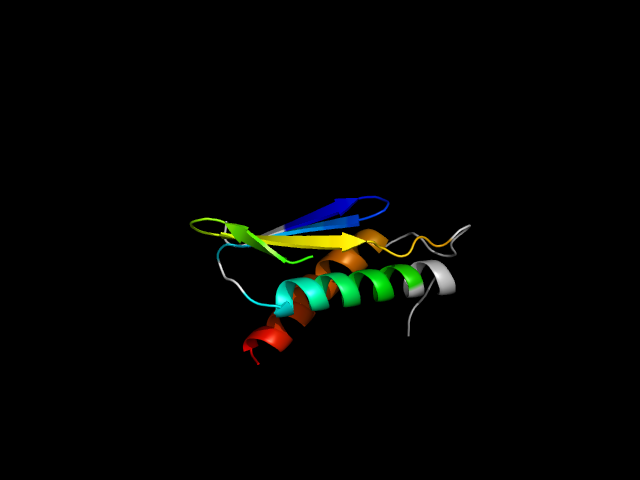 |
d1gc5a_ | A346-A389,A423-A467 | Alpha and beta proteins (a/b) | Ribokinase-like | Ribokinase-like | ADP-specific Phosphofructokinase/Glucokinase | ADP-dependent glucokinase |
1omh_Ac
 |
d1omha_ | A2-A132,A133-A293 | Alpha and beta proteins (a+b) | Origin of replication-binding domain, RBD-like | Origin of replication-binding domain, RBD-like | Relaxase domain | TrwC relaxase
|
Justification for analogy
comparing d1gc5a_ & d1rkd__ (other family, but sure homolog to d1gc5a_, since they are very similar, including an inserted domain). The insertion
that forms part of the DCoH motif is absent in d1rkd__. So the motif in d1gc5a_ is a hybrid.
In the alignments below, two upper-case letters are aligned, while two lower-case letters are NOT aligned. The two sequences in an alignment are NOT continuous in most cases (see their ranges). The superpositions are re-constructed from the corresponding alignments by minimizing the overall RMSD. In a PDB-format superposition file, the first domain is represented by chain S and the second domain is represented by chain A.
MANUAL alignment PDB format superposition aligned motifs in PyMol whole structures in PyMol
1gc5_Ah ieriHFHTY------------------------------------------------------------------GYYLALTQy----------------rGEEVRDALLFASLAAAakamkgnle---------------GLIDM----VDRQLAFVPTKIVaspk----------stvgIGDTISSSAFVSEFGMRKR---------------------------------------------------------------------------------------
1omh_Ac ----LSHMVltrqdigraasyygdasewqgkgaeelglsgevdskrfrellagnigeghrimrsatrqdskerigLDLTFSAPksvslqalvagdaeiikaHDRAVARTLEQAEARAqarqkiq--gktriettgnlvigkFRHETsrerDPQLHTHAVILNMtkrsdgqwralkndeivkATRYLGAVYNAELAHELQKlgyqlrygkdgnfdlahidrqqiegfskrteqiaewyaargldpnsvsleqkqaakvlsrakktsvdrealraewqatakelgidfs
DALI alignment PDB format superposition aligned motifs in PyMol whole structures in PyMol
1gc5_Ah ierihFHTY--------------------------------------------------------------------GYYLAL----TQYR--------GEEVRDALLFASLAAAAKAM-----------------kgnleGLIDM--VDRQLAFVPTKIVA---------SPKSTV--GIGDTISSSAFVSEFGMRKR---------------------------------------------------------------------------------------
1omh_Ac -----LSHMvltrqdigraasyygdasewqgkgaeelglsgevdskrfrellagnigeghrimrsatrqdskeriglDLTFSApksvSLQAlvagdaeiIKAHDRAVARTLEQAEARAQarqkiqgktriettgnlvigkfRHETSreRDPQLHTHAVILNMtkrsdgqwrALKNDEivKATRYLGAVYNAELAHELQKlgyqlrygkdgnfdlahidrqqiegfskrteqiaewyaargldpnsvsleqkqaakvlsrakktsvdrealraewqatakelgidfs
TM alignment PDB format superposition aligned motifs in PyMol whole structures in PyMol
1gc5_Ah IERIHFHT-----------------------------------------------------------------------YGYYLALT--QY---R-GEEVRDALLFASLAAAAKAMKG-------------------------NLEGLI----DMVDRQLAFVPTKIV-A-------------SPKS--TV-GIGDTISSSAFVSEFGMRK-------------------------------------------------------------------------------------R
1omh_Ac ------LSHMVLTRQDIGRAASYYGDASEWQGKGAEELGLSGEVDSKRFRELLAGNIGEGHRIMRSATRQDSKERIGLDLTFSAPKSVSLQALVAGDAEIIKAHDRAVARTLEQAEARAQARQKIQGKTRIETTGNLVIGKFR---HETSRER-----DPQLHTHAVILNMTKRSDGQWRALKNDEIVKATRYLGAVYNAELAHELQK-LGYQLRYGKDGNFDLAHIDRQQIEGFSKRTEQIAEWYAARGLDPNSVSLEQKQAAKVLSRAKKTSVDREALRAEWQATAKELGIDFS-
FAST alignment PDB format superposition aligned motifs in PyMol whole structures in PyMol
1gc5_Ah IERIHFHTYG--------------------------------------------------------------------------YYLALTQ---------YRGEEVRDALLFASLAAAAKAMKGNLEGLIDMVDR----------------------------------QLAFVPTKIVA-------------SPKS----TVGIGDTISSSAFVSEFGMRKR--------------------------------------------------------------------------------------
1omh_Ac ----------LSHMVLTRQDIGRAASYYGDASEWQGKGAEELGLSGEVDSKRFRELLAGNIGEGHRIMRSATRQDSKERIGLDLTFSAP--KSVSLQALVAGDAEIIKAHDRAVARTLEQAEA------------RAQARQKIQGKTRIETTGNLVIGKFRHETSRERDPQLHTH-AVILNMTKRSDGQWRALKNDEIVKATRYLGAVYNAELAHELQKL---GYQLRYGKDGNFDLAHIDRQQIEGFSKRTEQIAEWYAARGLDPNSVSLEQKQAAKVLSRAKKTSVDREALRAEWQATAKELGIDFS