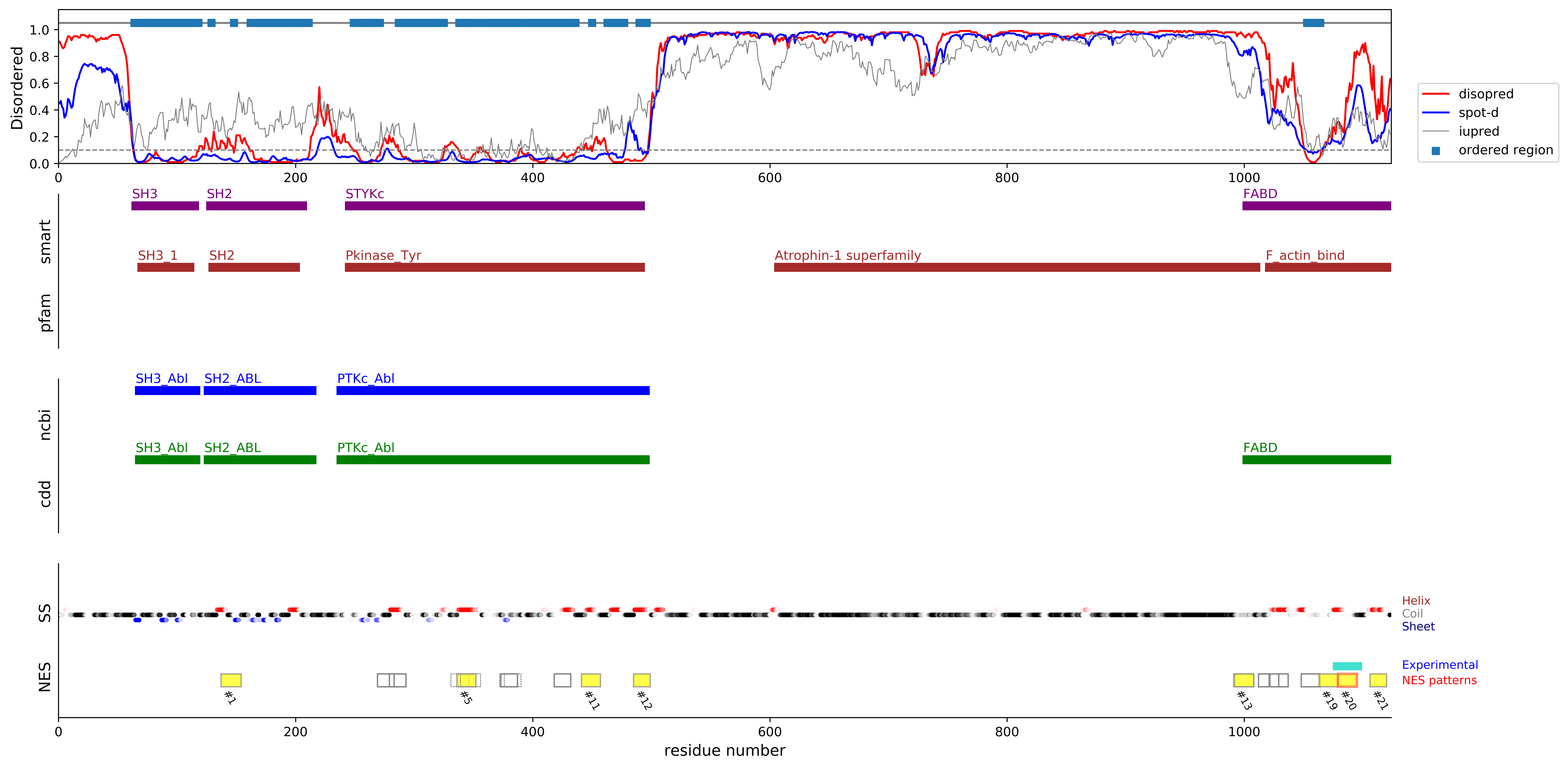
*Experimental: mutation (blue); functional region (cyan); located in the long functional sequence (lightblue)
*NES patterns: region with experimental evidence (red-orange-yellow); false positive region (gray)
| # | candidates | id | start# | sequence | secondary | class | multi-pattern | diso | spotd | iup | loc_DISO | loc_CDD | beta |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | fp_D | P00520 | 137 | AEYLLSSGINGSFLVRE | HHHHHCCCCCCCEEEEC | c1c-4 | uniq | 0.158 | 0.025 | 0.313 | boundary | boundary|SH3_Abl; boundary|SH2_ABL; | 0.0 |
| 2 | fp_D | P00520 | 269 | AVKTLKEDTMEVEE | EEEECCCCCCCHHH | c3-AT | uniq | 0.141 | 0.06 | 0.22 | boundary | MID|PTKc_Abl; | 0.143 |
| 3 | fp_D | P00520 | 279 | EVEEFLKEAAVMKE | CHHHHHHHHHHHHC | c3-AT | uniq | 0.12 | 0.044 | 0.235 | boundary | MID|PTKc_Abl; | 0.0 |
| 4 | fp_D | P00520 | 331 | NRQEVSAVVLLYMAT | CCCCCCHHHHHHHHH | c1b-4 | multi | 0.089 | 0.035 | 0.047 | boundary | MID|PTKc_Abl; | 0.0 |
| 5 | fp_D | P00520 | 336 | SAVVLLYMATQISSAM | CHHHHHHHHHHHHHHH | c1aR-4 | multi-selected | 0.055 | 0.014 | 0.044 | boundary | MID|PTKc_Abl; | 0.0 |
| 6 | fp_D | P00520 | 339 | VLLYMATQISSAMEYLE | HHHHHHHHHHHHHHHHH | c1cR-5 | multi | 0.055 | 0.012 | 0.054 | boundary | MID|PTKc_Abl; | 0.0 |
| 7 | fp_beta_O | P00520 | 372 | GENHLVKVADFGLSR | CCCCEEEECCCCCCC | c1b-4 | multi | 0.018 | 0.021 | 0.081 | ORD | MID|PTKc_Abl; | 0.571 |
| 8 | fp_beta_O | P00520 | 373 | ENHLVKVADFGLSR | CCCEEEECCCCCCC | c2-4 | multi-selected | 0.019 | 0.021 | 0.082 | ORD | MID|PTKc_Abl; | 0.571 |
| 9 | fp_O | P00520 | 376 | LVKVADFGLSRLMT | EEEECCCCCCCCCC | c3-AT | multi | 0.03 | 0.026 | 0.095 | ORD | MID|PTKc_Abl; | 0.143 |
| 10 | fp_D | P00520 | 418 | IKSDVWAFGVLLWE | CCCHHHHHHHHHHH | c2-AT-4 | uniq | 0.046 | 0.018 | 0.021 | boundary | MID|PTKc_Abl; | 0.0 |
| 11 | fp_D | P00520 | 441 | PGIDLSQVYELLEKDY | CCCCHHHHHHHHHHCC | c1aR-4 | uniq | 0.133 | 0.047 | 0.229 | boundary | MID|PTKc_Abl; | 0.0 |
| 12 | fp_D | P00520 | 485 | SFAEIHQAFETMFQ | CHHHHHHHHHHHHH | c3-4 | uniq | 0.046 | 0.123 | 0.423 | boundary | boundary|PTKc_Abl; | 0.0 |
| 13 | fp_D | P00520 | 991 | SSAAFIPLISTRVSLRK | CCCCCCCCCCCCCCCCC | c1c-4 | multi-selected | 0.963 | 0.841 | 0.539 | DISO | boundary|FABD; | 0.0 |
| 14 | fp_D | P00520 | 992 | SAAFIPLISTRVSLRK | CCCCCCCCCCCCCCCC | c1d-4 | multi | 0.962 | 0.837 | 0.535 | DISO | boundary|FABD; | 0.0 |
| 15 | fp_D | P00520 | 1012 | PERIASGTITKGVVLDS | CCCCCCCCCCHHHHHHH | c1c-AT-4 | uniq | 0.738 | 0.454 | 0.507 | DISO | boundary|FABD; | 0.0 |
| 16 | fp_D | P00520 | 1021 | TKGVVLDSTEALCLAI | CHHHHHHHHHHHHHHH | c1a-AT-4 | multi-selected | 0.579 | 0.357 | 0.353 | DISO | MID|FABD; | 0.0 |
| 17 | fp_D | P00520 | 1022 | KGVVLDSTEALCLAI | HHHHHHHHHHHHHHH | c1b-AT-4 | multi | 0.563 | 0.361 | 0.337 | DISO | MID|FABD; | 0.0 |
| 18 | fp_D | P00520 | 1048 | AVLEAGKNLYTFCVSY | HHHHHHCCCCCCCCCC | c1a-AT-5 | uniq | 0.061 | 0.096 | 0.209 | boundary | MID|FABD; | 0.0 |
| 19 | fp_D | P00520 | 1063 | YVDSIQQMRNKFAFRE + | CCCCCHHCCCCCHHHH | c1d-5 | uniq | 0.168 | 0.14 | 0.203 | boundary | MID|FABD; | 0.0 |
| 20 | cand_D | P00520 | 1079 | AINKLESNLRELQICP ++++++++++++++++ | HHHHHHHHCCCCCCCC | c1a-5 | uniq | 0.467 | 0.281 | 0.275 | DISO | MID|FABD; | 0.0 |
| 21 | fp_D | P00520 | 1106 | DFSKLLSSVKEISD | HHHHHHHHHHHHHH | c3-4 | uniq | 0.499 | 0.201 | 0.222 | DISO | boundary|FABD; | 0.0 |
*candidates: NES candidates and false positives annotated with "cand" and "fp", respectively;
if the segment is located in the disordered or boundary region, flagged with "D"; if the segment is located in the ordered region, flagged with "O";
if the segment's beta-strand content is over 0.5, flagged with "beta".
*sequence: Hydrophobic positions are colored in red. The positions with the experimental evidence is marked with '*' (mutation) and '+' (functional sequence in NESdb or sites in validNES).
The positions with '.' are for the region annotated in the long (more than 25 residues) functional sequence or site.
*multi-pattern: the consensus pattern is unique or multiple within the region (if the start# difference is less than 5, the segments are considered to be the same region)
*diso: average DISOPRED3-predicted disorder propensity for the segment
*spotd: average SPOT-Disorder-predicted disorder propensity for the segment
*iup: average IUPRED2A-predicted disorder propensity for the segment
*loc_DISO: location of the segment with respect to the ordered/disordered regions
*loc_CDD: location of the segment with respect to the conserved region annotated in the Conserved Domain Database (CDD)
*beta: beta-strand content in the middle of the segment