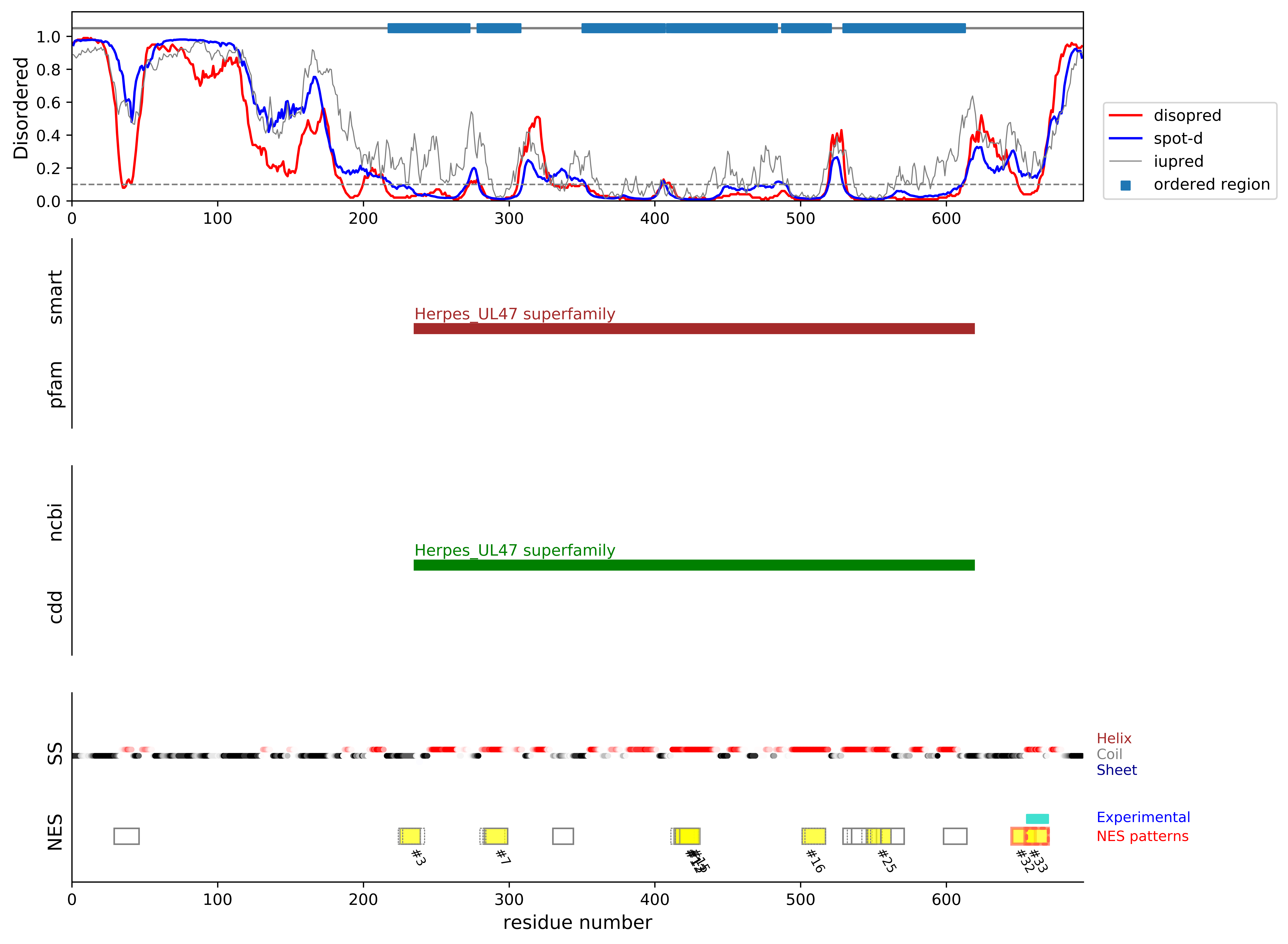
*Experimental: mutation (blue); functional region (cyan); located in the long functional sequence (lightblue)
*NES patterns: region with experimental evidence (red-orange-yellow); false positive region (gray)
| # | candidates | id | start# | sequence | secondary | class | multi-pattern | diso | spotd | iup | loc_DISO | loc_CDD | beta |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | fp_D | P10231 | 29 | DGVGFMGYLRAVFRGDD | CCCCCHHHHHHHHCCCC | c1cR-4 | uniq | 0.236 | 0.714 | 0.563 | DISO | 0.0 | |
| 2 | fp_D | P10231 | 224 | CPDSAFGLSRVGVMH | CCCCCCCCCCCCCCE | c1b-AT-4 | multi | 0.026 | 0.072 | 0.237 | boundary | boundary|Herpes_UL47 superfamily; | 0.0 |
| 3 | fp_D | P10231 | 225 | PDSAFGLSRVGVMH | CCCCCCCCCCCCCE | c2-4 | multi-selected | 0.026 | 0.071 | 0.239 | boundary | boundary|Herpes_UL47 superfamily; | 0.0 |
| 4 | fp_D | P10231 | 227 | SAFGLSRVGVMHFAS | CCCCCCCCCCCEECC | c1b-4 | multi | 0.029 | 0.061 | 0.221 | boundary | boundary|Herpes_UL47 superfamily; | 0.0 |
| 5 | fp_D | P10231 | 280 | PAQAMSFLADAVVRLAI | HHHHHHHHHHHHHHHHH | c1c-AT-4 | multi | 0.029 | 0.025 | 0.149 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 6 | fp_D | P10231 | 282 | QAMSFLADAVVRLAING | HHHHHHHHHHHHHHHHH | c1c-AT-4 | multi | 0.019 | 0.019 | 0.112 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 7 | fp_D | P10231 | 283 | AMSFLADAVVRLAING | HHHHHHHHHHHHHHHH | c1a-5 | multi-selected | 0.016 | 0.017 | 0.097 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 8 | fp_D | P10231 | 283 | AMSFLADAVVRLAING | HHHHHHHHHHHHHHHH | c1d-AT-5 | multi | 0.016 | 0.017 | 0.097 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 9 | fp_D | P10231 | 284 | MSFLADAVVRLAING | HHHHHHHHHHHHHHH | c1b-AT-4 | multi | 0.014 | 0.016 | 0.081 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 10 | fp_D | P10231 | 330 | ALRPVGAAAVPLLS | CCCCCCCCCCCCCC | c2-AT-5 | uniq | 0.089 | 0.163 | 0.116 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 11 | fp_D | P10231 | 411 | LTREAAFLGRVLDVLA | CHHHHHHHHHHHHHHH | c1d-AT-4 | multi | 0.029 | 0.016 | 0.039 | __ | MID|Herpes_UL47 superfamily; | 0.0 |
| 12 | fp_D | P10231 | 413 | REAAFLGRVLDVLAVMA | HHHHHHHHHHHHHHHHH | c1c-4 | multi-selected | 0.021 | 0.013 | 0.035 | __ | MID|Herpes_UL47 superfamily; | 0.0 |
| 13 | fp_D | P10231 | 414 | EAAFLGRVLDVLAVMA | HHHHHHHHHHHHHHHH | c1a-4 | multi-selected | 0.017 | 0.012 | 0.032 | __ | MID|Herpes_UL47 superfamily; | 0.0 |
| 14 | fp_D | P10231 | 414 | EAAFLGRVLDVLAVMA | HHHHHHHHHHHHHHHH | c1d-4 | multi | 0.017 | 0.012 | 0.032 | __ | MID|Herpes_UL47 superfamily; | 0.0 |
| 15 | fp_D | P10231 | 417 | FLGRVLDVLAVMAE | HHHHHHHHHHHHHH | c3-4 | multi-selected | 0.013 | 0.01 | 0.031 | __ | MID|Herpes_UL47 superfamily; | 0.0 |
| 16 | fp_D | P10231 | 501 | AVYALHTALATVTLKY | HHHHHHHHHHHHHHHH | c1a-5 | multi-selected | 0.014 | 0.017 | 0.051 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 17 | fp_D | P10231 | 501 | AVYALHTALATVTLKY | HHHHHHHHHHHHHHHH | c1d-AT-5 | multi | 0.014 | 0.017 | 0.051 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 18 | fp_D | P10231 | 503 | YALHTALATVTLKY | HHHHHHHHHHHHHH | c2-AT-4 | multi | 0.014 | 0.017 | 0.052 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 19 | fp_D | P10231 | 529 | DAAATRAILAAGLVLQR | HHHHHHHHHHHHHHHHH | c1c-AT-4 | multi-selected | 0.054 | 0.033 | 0.131 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 20 | fp_D | P10231 | 532 | ATRAILAAGLVLQR | HHHHHHHHHHHHHH | c2-AT-4 | multi | 0.019 | 0.022 | 0.095 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 21 | fp_D | P10231 | 535 | AILAAGLVLQRLLGFAD | HHHHHHHHHHHHHHHHH | c1c-AT-5 | multi-selected | 0.011 | 0.015 | 0.05 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 22 | fp_D | P10231 | 535 | AILAAGLVLQRLLG | HHHHHHHHHHHHHH | c3-AT | multi | 0.011 | 0.016 | 0.056 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 23 | fp_D | P10231 | 542 | VLQRLLGFADTVVA | HHHHHHHHHHHHHH | c3-AT | multi | 0.01 | 0.012 | 0.026 | boundary | MID|Herpes_UL47 superfamily; | 0.0 |
| 24 | fp_D | P10231 | 545 | RLLGFADTVVACVTLAA | HHHHHHHHHHHHHHHHH | c1cR-4 | multi | 0.012 | 0.014 | 0.035 | __ | MID|Herpes_UL47 superfamily; | 0.0 |
| 25 | fp_D | P10231 | 545 | RLLGFADTVVACVTLAA | HHHHHHHHHHHHHHHHH | c1c-5 | multi-selected | 0.012 | 0.014 | 0.035 | __ | MID|Herpes_UL47 superfamily; | 0.0 |
| 26 | fp_O | P10231 | 546 | LLGFADTVVACVTLAA | HHHHHHHHHHHHHHHH | c1a-AT-5 | multi | 0.012 | 0.014 | 0.036 | ORD | MID|Herpes_UL47 superfamily; | 0.0 |
| 27 | fp_O | P10231 | 546 | LLGFADTVVACVTLAA | HHHHHHHHHHHHHHHH | c1d-AT-5 | multi | 0.012 | 0.014 | 0.036 | ORD | MID|Herpes_UL47 superfamily; | 0.0 |
| 28 | fp_O | P10231 | 548 | GFADTVVACVTLAA | HHHHHHHHHHHHHH | c2-AT-5 | multi | 0.013 | 0.015 | 0.038 | ORD | MID|Herpes_UL47 superfamily; | 0.0 |
| 29 | fp_O | P10231 | 555 | ACVTLAAFDGGFTAPE | HHHHHHHHHCCCCCCC | c1aR-4 | uniq | 0.015 | 0.035 | 0.093 | ORD | MID|Herpes_UL47 superfamily; | 0.0 |
| 30 | fp_D | P10231 | 598 | FWADVRAAAEHVDLRP | HHHHHHHHHHHCCCCC | c1a-AT-4 | multi-selected | 0.05 | 0.073 | 0.32 | boundary | boundary|Herpes_UL47 superfamily; | 0.0 |
| 31 | fp_D | P10231 | 598 | FWADVRAAAEHVDLRP | HHHHHHHHHHHCCCCC | c1d-AT-4 | multi | 0.05 | 0.073 | 0.32 | boundary | boundary|Herpes_UL47 superfamily; | 0.0 |
| 32 | cand_D | P10231 | 645 | GGSVLGPRVRVVDIMS +++ | CCCCCCCCCCHHHHHH | c1a-4 | uniq | 0.079 | 0.195 | 0.287 | DISO | boundary|Herpes_UL47 superfamily; | 0.0 |
| 33 | cand_D | P10231 | 654 | RVVDIMSQFRKLLMGD ++++++++++ | CHHHHHHHHHHHHHCC | c1a-5 | multi-selected | 0.113 | 0.185 | 0.233 | DISO | boundary|Herpes_UL47 superfamily; | 0.0 |
| 34 | cand_D | P10231 | 655 | VVDIMSQFRKLLMGD ++++++++++ | HHHHHHHHHHHHHCC | c1b-5 | multi | 0.118 | 0.187 | 0.229 | DISO | 0.0 |
*candidates: NES candidates and false positives annotated with "cand" and "fp", respectively;
if the segment is located in the disordered or boundary region, flagged with "D"; if the segment is located in the ordered region, flagged with "O";
if the segment's beta-strand content is over 0.5, flagged with "beta".
*sequence: Hydrophobic positions are colored in red. The positions with the experimental evidence is marked with '*' (mutation) and '+' (functional sequence in NESdb or sites in validNES).
The positions with '.' are for the region annotated in the long (more than 25 residues) functional sequence or site.
*multi-pattern: the consensus pattern is unique or multiple within the region (if the start# difference is less than 5, the segments are considered to be the same region)
*diso: average DISOPRED3-predicted disorder propensity for the segment
*spotd: average SPOT-Disorder-predicted disorder propensity for the segment
*iup: average IUPRED2A-predicted disorder propensity for the segment
*loc_DISO: location of the segment with respect to the ordered/disordered regions
*loc_CDD: location of the segment with respect to the conserved region annotated in the Conserved Domain Database (CDD)
*beta: beta-strand content in the middle of the segment