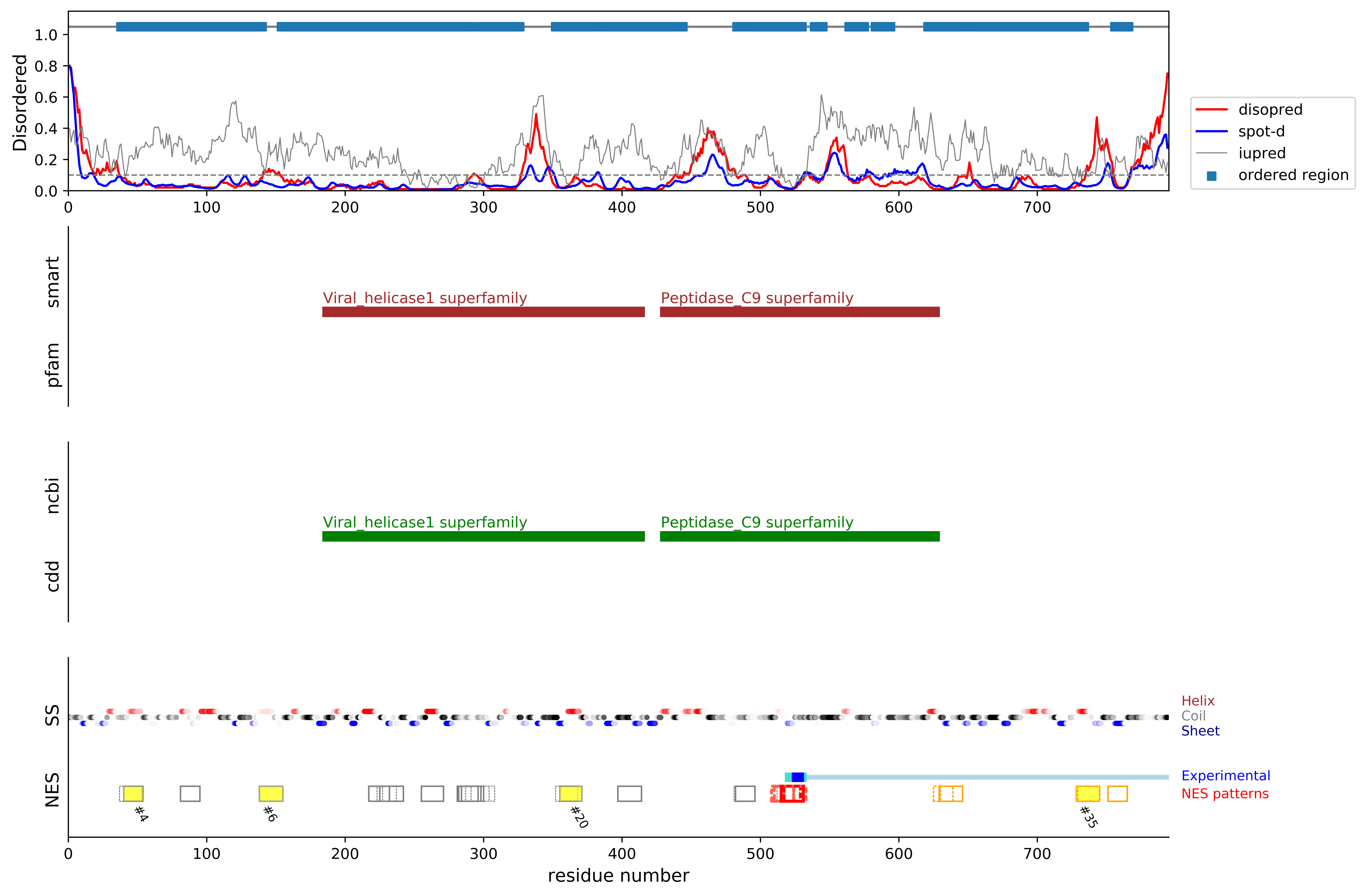
*Experimental: mutation (blue); functional region (cyan); located in the long functional sequence (lightblue)
*NES patterns: region with experimental evidence (red-orange-yellow); false positive region (gray)
| # | candidates | id | start# | sequence | secondary | class | multi-pattern | diso | spotd | iup | loc_DISO | loc_CDD | beta |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | fp_D | P36328 | 37 | KLSCIHPLAEQVIVITH | CCCCCCHHHHHHHHHHC | c1c-AT-5 | multi | 0.057 | 0.048 | 0.215 | boundary | 0.0 | |
| 2 | fp_D | P36328 | 37 | KLSCIHPLAEQVIVIT | CCCCCCHHHHHHHHHH | c1a-AT-5 | multi | 0.058 | 0.048 | 0.212 | boundary | 0.0 | |
| 3 | fp_D | P36328 | 37 | KLSCIHPLAEQVIVIT | CCCCCCHHHHHHHHHH | c1d-5 | multi | 0.058 | 0.048 | 0.212 | boundary | 0.0 | |
| 4 | fp_D | P36328 | 40 | CIHPLAEQVIVITH | CCCHHHHHHHHHHC | c3-4 | multi-selected | 0.05 | 0.041 | 0.204 | boundary | 0.0 | |
| 5 | fp_O | P36328 | 81 | DFQALSESATIVYN | HHHHHHCCCEEEEC | c3-AT | uniq | 0.031 | 0.028 | 0.243 | ORD | 0.143 | |
| 6 | fp_D | P36328 | 138 | RKQCVKKELVTGLGLTG | HHHHHHHHHHCCCCEEE | c1c-4 | uniq | 0.11 | 0.041 | 0.23 | boundary | 0.0 | |
| 7 | fp_O | P36328 | 217 | IIRDVKKMKGLDVNA | HHHHHHHCCCCCEEE | c1b-5 | uniq | 0.043 | 0.026 | 0.217 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 8 | fp_beta_O | P36328 | 223 | KMKGLDVNARTVDS | HCCCCCEEEEECCE | c3-AT | multi | 0.032 | 0.023 | 0.205 | ORD | MID|Viral_helicase1 superfamily; | 0.571 |
| 9 | fp_beta_O | P36328 | 225 | KGLDVNARTVDSVLLNG | CCCCEEEEECCEEECCC | c1c-AT-4 | multi-selected | 0.023 | 0.021 | 0.203 | ORD | MID|Viral_helicase1 superfamily; | 0.75 |
| 10 | fp_beta_O | P36328 | 227 | LDVNARTVDSVLLNG | CCEEEEECCEEECCC | c1b-AT-4 | multi | 0.018 | 0.018 | 0.195 | ORD | MID|Viral_helicase1 superfamily; | 0.714 |
| 11 | fp_O | P36328 | 255 | FACHAGTLRALIAIIR | CCCCHHHHHHHHHHHC | c1d-AT-4 | uniq | 0.01 | 0.006 | 0.057 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 12 | fp_O | P36328 | 281 | KQCGFFNMMCLKVHF | CCCCCCCCCCCCCCC | c1b-4 | multi | 0.06 | 0.042 | 0.038 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 13 | fp_O | P36328 | 281 | KQCGFFNMMCLKVHFNH | CCCCCCCCCCCCCCCCC | c1c-4 | multi-selected | 0.063 | 0.043 | 0.04 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 14 | fp_O | P36328 | 282 | QCGFFNMMCLKVHF | CCCCCCCCCCCCCC | c2-4 | multi-selected | 0.064 | 0.043 | 0.037 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 15 | fp_O | P36328 | 282 | QCGFFNMMCLKVHFNH | CCCCCCCCCCCCCCCC | c1d-4 | multi | 0.066 | 0.043 | 0.039 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 16 | fp_O | P36328 | 284 | GFFNMMCLKVHFNHEI | CCCCCCCCCCCCCCCC | c1aR-4 | multi-selected | 0.073 | 0.044 | 0.04 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 17 | fp_O | P36328 | 287 | NMMCLKVHFNHEICTQV | CCCCCCCCCCCCCCCCE | c1cR-4 | multi | 0.071 | 0.043 | 0.055 | ORD | MID|Viral_helicase1 superfamily; | 0.0 |
| 18 | fp_D | P36328 | 291 | LKVHFNHEICTQVFHKS | CCCCCCCCCCCCEEEEE | c1cR-4 | multi | 0.059 | 0.039 | 0.076 | boundary | MID|Viral_helicase1 superfamily; | 0.0 |
| 19 | fp_beta_D | P36328 | 352 | DDLILTCFRGWVKQLQ | CCEEEEEEHHHHHHHH | c1aR-5 | multi | 0.04 | 0.029 | 0.107 | boundary | MID|Viral_helicase1 superfamily; | 0.571 |
| 20 | fp_D | P36328 | 355 | ILTCFRGWVKQLQIDY | EEEEEHHHHHHHHHHC | c1a-5 | multi-selected | 0.049 | 0.028 | 0.096 | boundary | MID|Viral_helicase1 superfamily; | 0.143 |
| 21 | fp_O | P36328 | 397 | NENPLYAPTSEHVNVLL | CCCCCCCCCCCCEEEEE | c1c-AT-4 | uniq | 0.011 | 0.045 | 0.313 | ORD | boundary|Viral_helicase1 superfamily; boundary|Peptidase_C9 superfamily; | 0.0 |
| 22 | fp_D | P36328 | 481 | ALVPVLKTAGIDMTT | CCCCCCCCCCCCCCH | c1b-AT-5 | multi | 0.084 | 0.05 | 0.19 | boundary | MID|Peptidase_C9 superfamily; | 0.0 |
| 23 | fp_D | P36328 | 482 | LVPVLKTAGIDMTT | CCCCCCCCCCCCCH | c2-AT-5 | multi-selected | 0.08 | 0.051 | 0.194 | boundary | MID|Peptidase_C9 superfamily; | 0.0 |
| 24 | cand_D | P36328 | 508 | KAHSAEIVLNQLCVRF +++ | CCCCHHHHHCCEEEEE | c1a-AT-4 | multi | 0.034 | 0.032 | 0.108 | boundary | MID|Peptidase_C9 superfamily; | 0.0 |
| 25 | cand_D | P36328 | 508 | KAHSAEIVLNQLCVRF +++ | CCCCHHHHHCCEEEEE | c1d-AT-4 | multi | 0.034 | 0.032 | 0.108 | boundary | MID|Peptidase_C9 superfamily; | 0.0 |
| 26 | cand_D | P36328 | 510 | HSAEIVLNQLCVRF +++ | CCHHHHHCCEEEEE | c2-4 | multi | 0.029 | 0.027 | 0.089 | boundary | MID|Peptidase_C9 superfamily; | 0.143 |
| 27 | fp_beta_D | P36328 | 512 | AEIVLNQLCVRFFGLD +++++*+ | HHHHHCCEEEEECCCC | c1aR-5 | multi | 0.024 | 0.025 | 0.073 | boundary | MID|Peptidase_C9 superfamily; | 0.571 |
| 28 | cand_beta_D | P36328 | 515 | VLNQLCVRFFGLDLDS +++++*+*++ | HHCCEEEEECCCCCCC | c1a-5 | multi-selected | 0.024 | 0.031 | 0.065 | boundary | MID|Peptidase_C9 superfamily; | 0.714 |
| 29 | cand_beta_D | P36328 | 515 | VLNQLCVRFFGLDL +++++*+* | HHCCEEEEECCCCC | c2-5 | multi | 0.019 | 0.025 | 0.064 | boundary | MID|Peptidase_C9 superfamily; | 0.714 |
| 30 | cand_D | P36328 | 517 | NQLCVRFFGLDLDSGL +++++*+*++.. | CCEEEEECCCCCCCCC | c1aR-4 | multi | 0.031 | 0.042 | 0.062 | boundary | MID|Peptidase_C9 superfamily; | 0.429 |
| 31 | cand_beta_D | P36328 | 517 | NQLCVRFFGLDLDS +++++*+*++ | CCEEEEECCCCCCC | c2-4 | multi | 0.024 | 0.032 | 0.057 | boundary | MID|Peptidase_C9 superfamily; | 0.571 |
| 32 | fp_D | P36328 | 625 | FVSKLKGRTVLVVG .............. | HHHHCCCCEEEEEE | c3-AT | multi | 0.011 | 0.022 | 0.233 | boundary | boundary|Peptidase_C9 superfamily; | 0.286 |
| 33 | fp_beta_D | P36328 | 629 | LKGRTVLVVGEKLSVPG ................. | CCCCEEEEEECCCCCCC | c1c-AT-4 | multi-selected | 0.032 | 0.023 | 0.224 | boundary | boundary|Peptidase_C9 superfamily; | 0.75 |
| 34 | fp_beta_D | P36328 | 630 | KGRTVLVVGEKLSVPG ................ | CCCEEEEEECCCCCCC | c1d-4 | multi | 0.034 | 0.023 | 0.225 | __ | boundary|Peptidase_C9 superfamily; | 0.714 |
| 35 | fp_D | P36328 | 728 | ASESIIGAIARQFKFSR ................. | CHHHHHHHHHHCEEEEE | c1c-4 | multi-selected | 0.159 | 0.035 | 0.126 | boundary | 0.0 | |
| 36 | fp_D | P36328 | 729 | SESIIGAIARQFKFSR ................ | HHHHHHHHHHCEEEEE | c1a-AT-4 | multi | 0.167 | 0.034 | 0.128 | boundary | 0.0 | |
| 37 | fp_D | P36328 | 729 | SESIIGAIARQFKFSR ................ | HHHHHHHHHHCEEEEE | c1d-4 | multi | 0.167 | 0.034 | 0.128 | boundary | 0.0 | |
| 38 | fp_beta_D | P36328 | 751 | SHEETEVLFVFIGY .............. | CCCCCEEEEEEEEE | c2-AT-4 | uniq | 0.091 | 0.055 | 0.158 | boundary | 0.714 |
*candidates: NES candidates and false positives annotated with "cand" and "fp", respectively;
if the segment is located in the disordered or boundary region, flagged with "D"; if the segment is located in the ordered region, flagged with "O";
if the segment's beta-strand content is over 0.5, flagged with "beta".
*sequence: Hydrophobic positions are colored in red. The positions with the experimental evidence is marked with '*' (mutation) and '+' (functional sequence in NESdb or sites in validNES).
The positions with '.' are for the region annotated in the long (more than 25 residues) functional sequence or site.
*multi-pattern: the consensus pattern is unique or multiple within the region (if the start# difference is less than 5, the segments are considered to be the same region)
*diso: average DISOPRED3-predicted disorder propensity for the segment
*spotd: average SPOT-Disorder-predicted disorder propensity for the segment
*iup: average IUPRED2A-predicted disorder propensity for the segment
*loc_DISO: location of the segment with respect to the ordered/disordered regions
*loc_CDD: location of the segment with respect to the conserved region annotated in the Conserved Domain Database (CDD)
*beta: beta-strand content in the middle of the segment