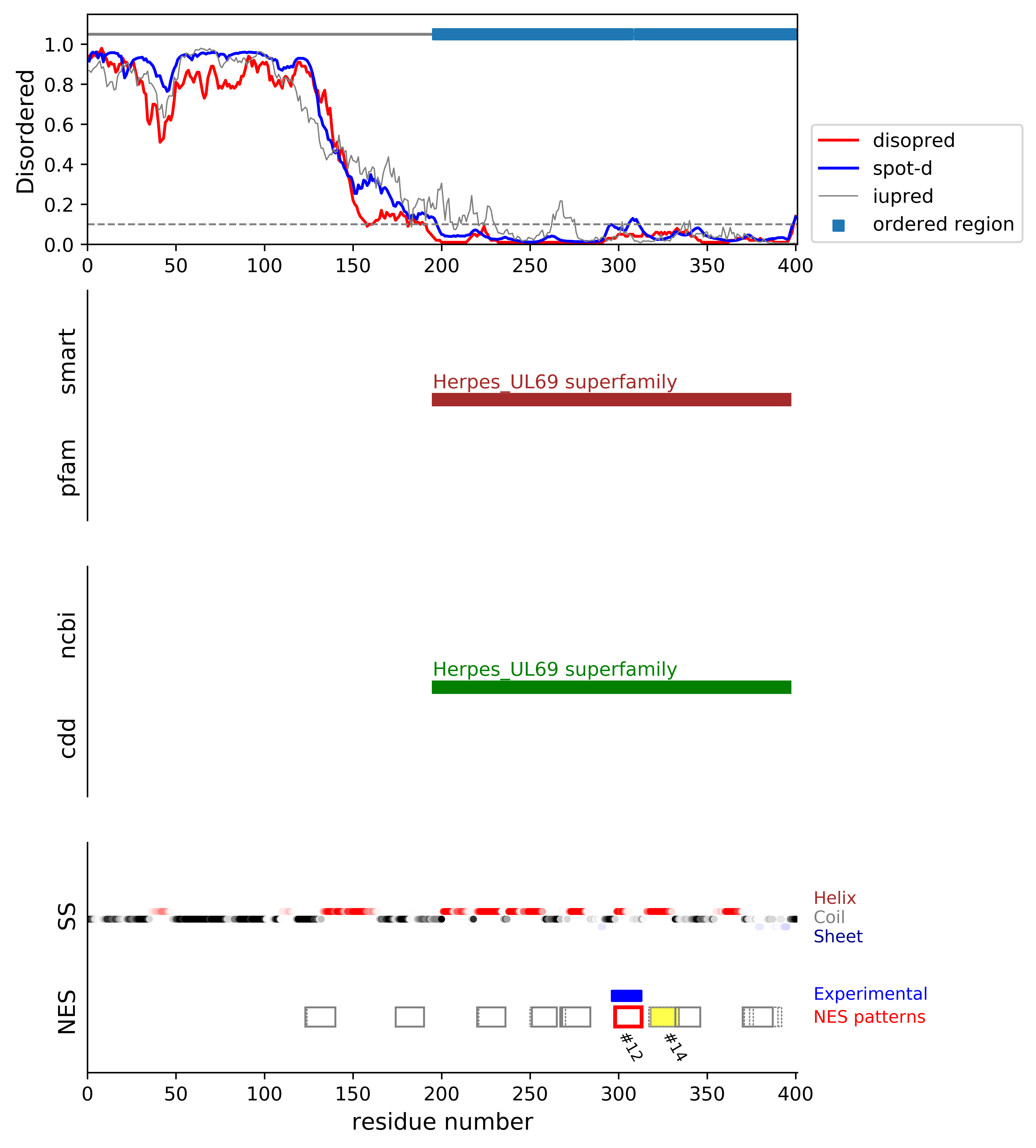
*Experimental: mutation (blue); functional region (cyan); located in the long functional sequence (lightblue)
*NES patterns: region with experimental evidence (red-orange-yellow); false positive region (gray)
| # | candidates | id | start# | sequence | secondary | class | multi-pattern | diso | spotd | iup | loc_DISO | loc_CDD | beta |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | fp_D | Q01354 | 123 | GGGAAARSIGSSLRLAR | CCCCCCCCCCHHHHHHH | c1c-AT-4 | multi-selected | 0.746 | 0.697 | 0.543 | DISO | 0.0 | |
| 2 | fp_D | Q01354 | 124 | GGAAARSIGSSLRLAR | CCCCCCCCCHHHHHHH | c1d-AT-4 | multi | 0.737 | 0.683 | 0.534 | DISO | 0.0 | |
| 3 | fp_D | Q01354 | 174 | RAAGVSPWAAVLDFGA | CCCCCCCCCHHCCCCC | c1a-AT-4 | uniq | 0.123 | 0.163 | 0.221 | boundary | boundary|Herpes_UL69 superfamily; | 0.0 |
| 4 | fp_O | Q01354 | 220 | HAAQAARALRDLVLRS | HHHHHHHHHHHHHHHC | c1a-AT-4 | multi-selected | 0.035 | 0.037 | 0.11 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 5 | fp_O | Q01354 | 221 | AAQAARALRDLVLRS | HHHHHHHHHHHHHHC | c1b-AT-4 | multi | 0.035 | 0.035 | 0.107 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 6 | fp_O | Q01354 | 250 | TWCKFIATKNLRLRT | HHHHHHHHCCCCCCC | c1b-AT-4 | multi | 0.01 | 0.024 | 0.059 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 7 | fp_O | Q01354 | 251 | WCKFIATKNLRLRT | HHHHHHHCCCCCCC | c2-AT-4 | multi-selected | 0.01 | 0.025 | 0.062 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 8 | fp_O | Q01354 | 267 | PIVATAGAVLENLRLKL | CHHCCHHHHHHHHHHHC | c1c-AT-5 | multi-selected | 0.01 | 0.015 | 0.091 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 9 | fp_O | Q01354 | 268 | IVATAGAVLENLRLKL | HHCCHHHHHHHHHHHC | c1a-AT-5 | multi-selected | 0.01 | 0.015 | 0.084 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 10 | fp_O | Q01354 | 268 | IVATAGAVLENLRLKL | HHCCHHHHHHHHHHHC | c1d-AT-5 | multi | 0.01 | 0.015 | 0.084 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 11 | fp_O | Q01354 | 270 | ATAGAVLENLRLKL | CCHHHHHHHHHHHC | c2-AT-4 | multi | 0.01 | 0.014 | 0.07 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 12 | cand_D | Q01354 | 298 | SLEELCAARRLSLAT *++*+++++*+* | CHHHHHHHHCCCCCC | c1b-AT-5 | uniq | 0.044 | 0.095 | 0.042 | boundary | MID|Herpes_UL69 superfamily; | 0.0 |
| 13 | fp_D | Q01354 | 317 | YMFVMLARLSRAVRSGA | HHHHHHHHHHHHHHCCC | c1cR-4 | multi | 0.055 | 0.036 | 0.026 | __ | MID|Herpes_UL69 superfamily; | 0.0 |
| 14 | fp_D | Q01354 | 318 | MFVMLARLSRAVRSGA | HHHHHHHHHHHHHCCC | c1aR-4 | multi-selected | 0.056 | 0.035 | 0.027 | __ | MID|Herpes_UL69 superfamily; | 0.0 |
| 15 | fp_O | Q01354 | 332 | GAECVPLLEVTVGD | CCCCCCCCCCCCCC | c2-4 | uniq | 0.053 | 0.06 | 0.066 | ORD | MID|Herpes_UL69 superfamily; | 0.0 |
| 16 | fp_O | Q01354 | 370 | ACDSMTCKLVANFTLVP | HCCCCCCCCEECCCCCC | c1c-4 | multi-selected | 0.023 | 0.029 | 0.025 | ORD | boundary|Herpes_UL69 superfamily; | 0.25 |
| 17 | fp_O | Q01354 | 371 | CDSMTCKLVANFTLVP | CCCCCCCCEECCCCCC | c1a-AT-4 | multi | 0.023 | 0.029 | 0.024 | ORD | boundary|Herpes_UL69 superfamily; | 0.286 |
| 18 | fp_O | Q01354 | 371 | CDSMTCKLVANFTLVP | CCCCCCCCEECCCCCC | c1d-AT-4 | multi | 0.023 | 0.029 | 0.024 | ORD | boundary|Herpes_UL69 superfamily; | 0.286 |
| 19 | fp_O | Q01354 | 374 | MTCKLVANFTLVPVYMHG | CCCCCEECCCCCCCEEEC | c4-4 | multi | 0.018 | 0.028 | 0.016 | ORD | boundary|Herpes_UL69 superfamily; | 0.25 |
| 20 | fp_O | Q01354 | 376 | CKLVANFTLVPVYM | CCCEECCCCCCCEE | c2-AT-4 | multi | 0.019 | 0.025 | 0.015 | ORD | boundary|Herpes_UL69 superfamily; | 0.286 |
*candidates: NES candidates and false positives annotated with "cand" and "fp", respectively;
if the segment is located in the disordered or boundary region, flagged with "D"; if the segment is located in the ordered region, flagged with "O";
if the segment's beta-strand content is over 0.5, flagged with "beta".
*sequence: Hydrophobic positions are colored in red. The positions with the experimental evidence is marked with '*' (mutation) and '+' (functional sequence in NESdb or sites in validNES).
The positions with '.' are for the region annotated in the long (more than 25 residues) functional sequence or site.
*multi-pattern: the consensus pattern is unique or multiple within the region (if the start# difference is less than 5, the segments are considered to be the same region)
*diso: average DISOPRED3-predicted disorder propensity for the segment
*spotd: average SPOT-Disorder-predicted disorder propensity for the segment
*iup: average IUPRED2A-predicted disorder propensity for the segment
*loc_DISO: location of the segment with respect to the ordered/disordered regions
*loc_CDD: location of the segment with respect to the conserved region annotated in the Conserved Domain Database (CDD)
*beta: beta-strand content in the middle of the segment