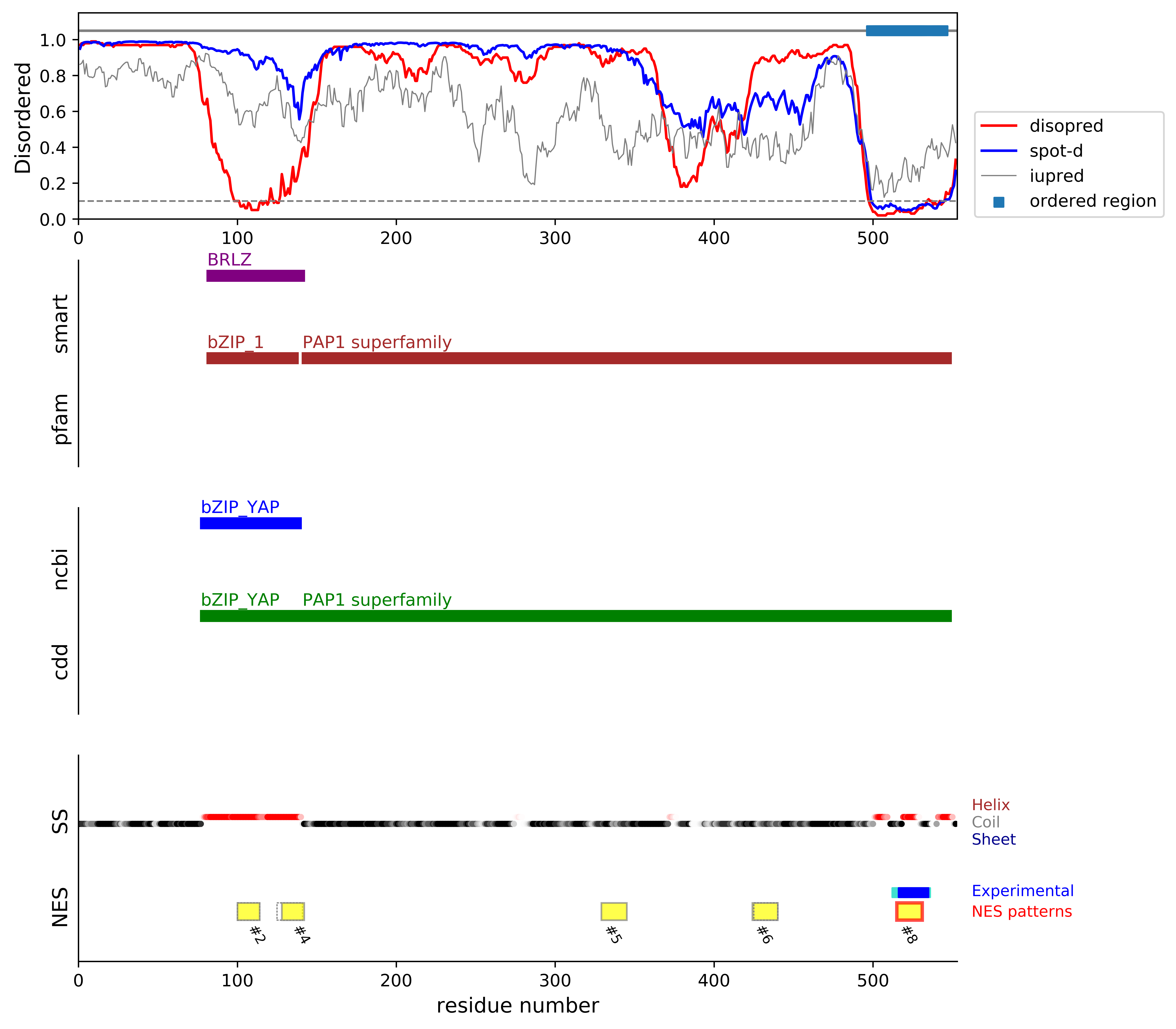
*Experimental: mutation (blue); functional region (cyan); located in the long functional sequence (lightblue)
*NES patterns: region with experimental evidence (red-orange-yellow); false positive region (gray)
| # | candidates | id | start# | sequence | secondary | class | multi-pattern | diso | spotd | iup | loc_DISO | loc_CDD | beta |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | fp_D | Q01663 | 100 | HLKALETQVVTLKE | HHHHHHHHHHHHHH | c2-AT-5 | multi | 0.069 | 0.898 | 0.573 | DISO | MID|bZIP_YAP; | 0.0 |
| 2 | fp_D | Q01663 | 100 | HLKALETQVVTLKE | HHHHHHHHHHHHHH | c3-4 | multi-selected | 0.069 | 0.898 | 0.573 | DISO | MID|bZIP_YAP; | 0.0 |
| 3 | fp_D | Q01663 | 125 | LRQKVRQLEEELRILK | HHHHHHHHHHHHHHHH | c1d-4 | multi | 0.196 | 0.73 | 0.572 | DISO | boundary|bZIP_YAP; boundary|PAP1 superfamily; | 0.0 |
| 4 | fp_D | Q01663 | 128 | KVRQLEEELRILKD | HHHHHHHHHHHHHC | c3-4 | multi-selected | 0.23 | 0.707 | 0.527 | DISO | boundary|bZIP_YAP; boundary|PAP1 superfamily; | 0.0 |
| 5 | fp_D | Q01663 | 329 | VSKSIPNVELSLNVNQ | CCCCCCCCCCCCCCCC | c1d-4 | uniq | 0.899 | 0.944 | 0.433 | DISO | MID|PAP1 superfamily; | 0.0 |
| 6 | fp_D | Q01663 | 424 | NGDLITNSLHGLDFLE | CCCCCCCCCCCCCCCC | c1a-4 | multi-selected | 0.883 | 0.666 | 0.418 | DISO | MID|PAP1 superfamily; | 0.0 |
| 7 | fp_D | Q01663 | 425 | GDLITNSLHGLDFLE | CCCCCCCCCCCCCCC | c1b-AT-4 | multi | 0.887 | 0.667 | 0.419 | DISO | MID|PAP1 superfamily; | 0.0 |
| 8 | cand_D | Q01663 | 515 | ESFDIDDLCSKLKNKA ++++*++*+++*++++ | CCCCHHHHHHHHHHHC | c1aR-4 | uniq | 0.043 | 0.057 | 0.256 | boundary | boundary|PAP1 superfamily; | 0.0 |
*candidates: NES candidates and false positives annotated with "cand" and "fp", respectively;
if the segment is located in the disordered or boundary region, flagged with "D"; if the segment is located in the ordered region, flagged with "O";
if the segment's beta-strand content is over 0.5, flagged with "beta".
*sequence: Hydrophobic positions are colored in red. The positions with the experimental evidence is marked with '*' (mutation) and '+' (functional sequence in NESdb or sites in validNES).
The positions with '.' are for the region annotated in the long (more than 25 residues) functional sequence or site.
*multi-pattern: the consensus pattern is unique or multiple within the region (if the start# difference is less than 5, the segments are considered to be the same region)
*diso: average DISOPRED3-predicted disorder propensity for the segment
*spotd: average SPOT-Disorder-predicted disorder propensity for the segment
*iup: average IUPRED2A-predicted disorder propensity for the segment
*loc_DISO: location of the segment with respect to the ordered/disordered regions
*loc_CDD: location of the segment with respect to the conserved region annotated in the Conserved Domain Database (CDD)
*beta: beta-strand content in the middle of the segment