| ProSMoS Home | Pattern Search by Matrix | Pattern Search by Structure | Examples | Help |
How to use ProSMoS Server
3. Instruction to define query structure pattern
4. Guild for motif search by matrix
5. Guild for motif search by protein structure
Introduction: Assessing structural similarity and defining common regions through comparison of protein spatial structures is an important task in functional and evolutionary studies of proteins. There are many servers that compare structures and define sub-structures in common between proteins through superposition and closeness of either coordinates or contacts. However, a natural way to analyze a structure for experts who works on structure classification is to look for specific three-dimensional motifs and patterns instead of finding common features in two proteins. Such motifs can be described by the architecture and topology of major secondary structural elements (SSEs) without consideration of subtle differences in 3D coordinates. Despite the importance of motif-based structure searches, currently there is only one web server TOPS devoted to this specific task; moreover, TOPS falls short since it finds topological match but ignores other important spatial properties, such as interactions and chirality. Recently, we developed an approach for protein structure pattern search, ProSMoS (ref below). Here, we implemented ProSMoS as a web-server. ProSMoS converts 3D structure into an interaction matrix representation including the SSE types, coordinates of SSE starts and ends, types of interactions between SSEs, b-sheet definitions and handedness. For a user-defined structure pattern, ProSMoS lists all structures from a database that contain this pattern. ProSMoS server will be of interest to structural biologists who are interested in structure motif searching and would like to analyze very general and distant structural similarities.
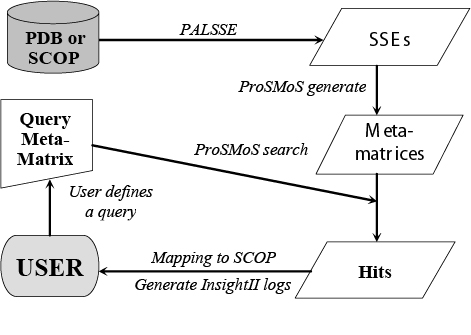
Processing method: ProSMoS server finds the structural pattern through the following steps: 1) pre-process each structure in PDB or ASTRAL databases to generate SSEs; 2) convert 3D structures into interaction matrix representations and store the results as a database of meta-matrices; 3) Search the database with a user-defined structure pattern; 4) List all the proteins in the database matching the query structure pattern. In addition to these basic functions, ProSMoS server maps the hits to SCOP database on different SCOP hierarchy level; and provides PyMOL script files to visualize the results on the fly.
Web address: http://prodata.swmed.edu/ProSMoS/
Input and Output: ProSMoS server runs in two modes: 1) motif search starting from a user-defined interaction matrix; and 2) motif search with a PDB for a protein structure. In the first mode, the input query matrix should be designed by user based on the set of rules we explain on the web site. In the second mode, user can input PDB ID, SCOP ID or upload a structure file in PDB format. The server will generate the interaction matrix from the structure. This matrix can be used "as is" to initiate searches, but the option is provided to edit the matrix. It is frequently beneficial to refine the query matrix by removing the uninteresting SSEs, revising the sheet and handedness information or editing the interactions between SSEs. Output is the list of proteins in PDB or SCOP matching the query motif. SCOP hierarchy information is given for proteins in SCOP and hits can be visualized on the fly with PyMOL.
Reference: S. Shi, Y. Zhong, I.Majumdar, S. Sri Krishna, and N.V. Grishin (2007). Searching for three-dimensional secondary structural patterns in proteins with ProSMoS. Bioinformatics, 23, 1331-1338.
2. Workingflow flowchart of ProSMoS:
3. Instruction for users to define query structure motif
Here, we define structure motif or structure pattern as the common region among different protein structures which means the same major secondary structures in similar spatial arrangement and with the same topological connections without consideration of subtle differences in three dimensional (3D) coordinates. ProSMoS operate on the 3D packing of secondary structure elements (SSEs).
For one specific structure motif, user can define how many major SSEs should be considered as the core of that motif and then define the interaction relationship between each pair SSEs in one matrix (interaction matrix) by following parameters:
X - presence or absence of interaction between two SSEs is not checked and not used;
x - interaction should be present, but the angles are not checked and not used;
C - interaction present (with or without H-bonds), the angle is less than 85 degree;
T - interaction present (with or without H-bonds), the angle is no less than 95 degree;
c - H-bond interaction present, the two b-strands are parallel;
t - H-bond interaction present, the two b-strands are antiparallel;
u - interaction present (without H-bonds), the angle is less than 85 degree;
v - interaction present (without H-bonds), the angle is no less than 95 degree;
N - interaction present, the angle is no less than 85 degree, but less than 95 degree;
- - no interaction present.
As the first time user, you may be confused about those parameters, so please refer the figure below:
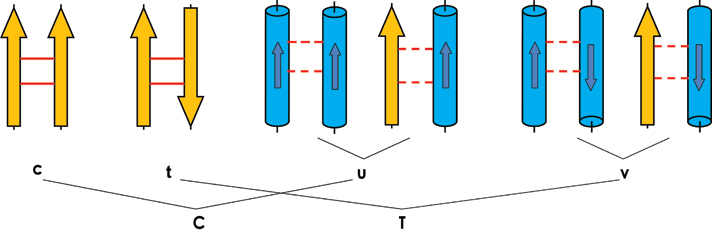
In the above figure, yellow arrow strands for beta strand; blue cylinder strands for helix and the smaller arrow inside the cylinder strands for the direction of helix. Solid line between two strands means H-bonding. Dash line between any type SSEs means interaction excepte H-bonding. Besides those 6 symbols shown in above figure, "−" is used for one pair SSEs without any interaction. "N" is used for there is interaction between two SSEs but the angel between them is not smaller than 85 degree and smaller than 95 degree. "X" is used for the case that you do not care if there is interaction between SSE pair or not, i.e. mapping {C,T,N, −}."x" is used for the case that you care the interaction but do not care the angel between SSE pair, i.e mapping {C,T,N}.
By using those ten symbols {c,t,u,v,C,T,N,−,X,x}, we can define the interaction matrix for structure motif or pattern. The interaction matrix is a upper triangle matrix looks like below:
1 2 3
E H E
* v c
* v
*
We can see, in the matrix, the first line is the index line list the how many SSEs presented in motif of your interest (starting from 1 to N); the second line lists the type of SSE ( H is a helical conformation, which includes all types of helices. E is a beta-strand, and L is a linker between two consecutive parallel beta-strands. The linker is usually a stretch of polypeptide chain in extended conformation, which does not have a neighbor to form hydrogen bonds with, and thus cannot be called a beta-strand. We introduced the linker element, because it is important for the topology. Two parallel beta-strands, consecutive in sequence, require a connection between them, and this connection is sometimes supplied by a loop. This loop is recorded in the meta-matrix as a linker L.); after the first two lines, there is the interaction matrix which is a upper triangle matrix with '*' in the main diagonal.
Sometimes, interaction matrix is not enough for deciphering the topology of motif. For example:
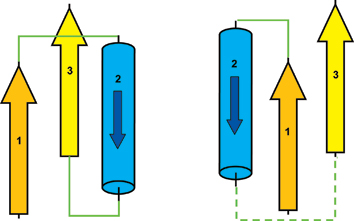
For interaction matrix shown above, we may get two different beta-alpha-beta topology. In the figure shown above, left beta-alpha-beta unit has right handedness as 1 2 3 R but right beta-alpha-beta unit has left handedness as 1 2 3 L. The only difference for those two units are the topological connections, i.e. the handedness.
So we provides handedness option when defining query motif. For example, for the left figure, we can write the query matrix as:
1 2 3
E H E
* v c
* v
*
handedness 1 2 3 R
The handedness line will specify right-handedness (R) or left-handedness (L) for selected elements. E.g. handedness 1 2 3 R symbolizes that the three elements 1,2, and 3 are right-handed.
We also provides several other options which may help user refine their query matrix for specific motif of interest:
sheets S, or sheet D: specifies beta-strands that are in the same beta-sheet
(sheet S) or in different beta-sheets (sheet D). E.g. sheet D 1 2 3 means that
beta-strands 1, 2, and 3 are not all in the same beta-sheet. It is OK if two of
these beta-strands is in the same sheet, but not all three.
chain S, or chain D: specified elements are in the same chain (chain S) or in
different chains (chain D); E.g. chain S 1 2 3 means that all three elements 1,
2, and 3 belong to a single PDB chain.
length: specify restrictions on the element length. E.g. length 3 H 8 50
requires the third element, which is an alpha-helix, to be at least 8 residues
long and no more than 50 residues long (8 and 50 are included).
parallel and antiparallel: specify the relationship of beta-strands that are in
the same beta-sheet, but are not hydrogen-bonded neighbors. E.g. parallel 1 3.
means that non-H-bonded beta-strands 1 and 3 are parallel.
Below we give one example for defining beta-grasp motif:
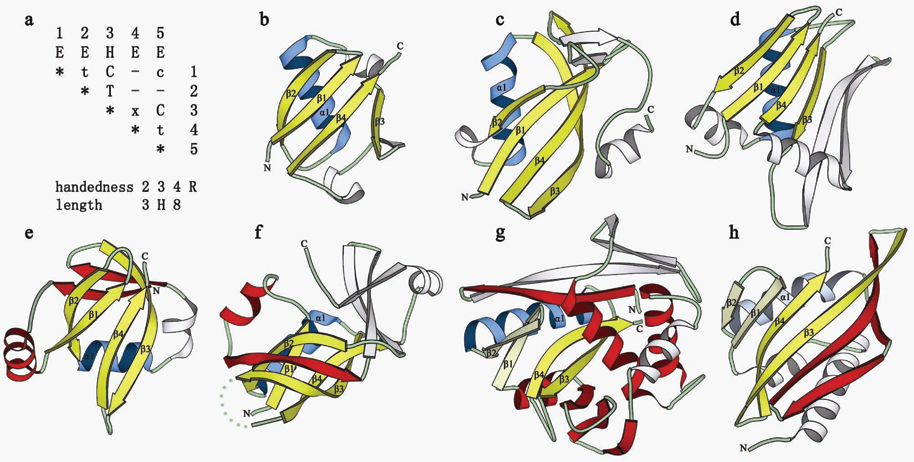
Panel (a) in above figure is query matrix we designed for beta-grasp fold:
1 2 3 4 5
E E H E E
* t C - c
* T - -
* x C
* t
*
handedness 2 3 4 R
length 3 H 8 100
Panels (b) to (h) show the motifs identified in different structures. Panel (b)
,(c) and (d) show the domain with beta-Grasp core; panel (e) and (f) show
beta-Grasp motif forms part of the core ; panel (g) and (h) shown beta-Grasp
motif partially overlaps with the core, driven by structure drift. beta-Strands
shown in yellow and beta-helices shown in blue comprise the core of the beta-Grasp
motif. These core elements are labeled. Non-beta-Grasp structural core SSEs are
colored red. Non-core SSEs are white. beta-Grasp structural core SSEs that are
insertions into the domain core (i.e. beta-Grasp formed by structural drift) are
shown in lighter colors (hairpin beta 1 beta 2 in g and h, and helix in h).
Want to see the output of beta-grasp pattern search? Please click here. Want to see more examples of pattern design? Please go to the examples page.
For detail of the analysis of beta-grasp motif, please refer our paper:
Shuoyong Shi, Yi Zhong, Indraneel Majumdar, Subramanian Sri Krishna, and Nick V. Grishin (2007). Searching for three-dimensional secondary structural patterns in proteins with ProSMoS. Bioinformatics, 23, 1331-1338.
4. User guild for motif search by matrix
ProSMoS server provides two different way to search motif against structure database. In this section, I would like introduce how to do motif search by matrix.
In the page: http://prodata.swmed.edu/ProSMoS/patternsearch.php
You will find a text window in which you can input query matrix for motif of interest.
If you want to know how to design query matrix and see some example, please refer the section 3.Instruction for users to define query structure motif. Here, I suppose you have grasped the rule of query matrix design.
You can copy and paste the query matrix you designed in the text window or upload the file that contains the query matrix.
You can choose the structure database you want to search against with.
Databases stored in our server are: PDB, SCOP, SCOP 40 and SCOP 95. We pre-calculated the secondary structure element assignment for each structure in those database and converted the 3D structure information into 2D meta-matrix. Those meta-matrices are then stored as databases in our server.
Here, I strongly recommend you input email address to receive results since our server also has other services provided which means that it is high loaded sometimes and it may need time for your job to wait in the que. So please input your email address and then when your job is done, an automatic email will be sent to you with the result link presented.
After submitting your query matrix, the server will check the correctness of your input. If there is some errors, a error message will show up, you can edit it again.
When query matrix passes the checking step, it will be used to search against the database you selected and then the hits will be mapped to SCOP on fold and superfamily level. The PyMOL scripts for each matched motif in protein structure will also be provided for visualization. You can also download the original result of ProSMoS in .tar format. Want to see the example output? Please click here.
5. User guild for motif search by protein structure
ProSMoS is originally designed to work in a supervised manner which means that user has some pre-knowledge about the structure feature of the motif of interest. If so, user can delineate those features by query matrix using the parameter we provided. However, for users who are not very familiar with common feature of the motif they studied or they can not design the query matrix directly, we provide another way to run ProSMoS server: user provide a protein structure, we provided a interactive way for help user to extract the important feature of that structure, design and refine corresponding query matrix. This matrix can be used "as is" to initiate searches, but the option is provided to edit the matrix. It is frequently beneficial to refine the query matrix by removing the uninteresting SSEs, revising the sheet and handedness information or editing the interactions between SSEs.
In the page: http://prodata.swmed.edu/ProSMoS/patterndesign.php
Three different way are provided for user to input protein structure, please select one way one time.
1). PDB ID plus range, you can input pdb entry code plus chain and the range in that chain. We will try to locate the corresponding pdb in our local server (our server update the PDB database weekly), and cut the chain and atom range you selected out. If you do not want to specify the chain and range info, just leave it blank. If you want to specify the atom range, please also input the corresponding chain, if there is no chain info in that PDB file, please use symbol "-".
2) SCOP ID, you can input SCOP sid which is a 7 character ID, for example d16pka_,d1a04a1
3) upload PDB file
For the user defined option shown in this page:
1) Minimal helix length: It is the length cutoff for selecting helix in structure, the helix with length < the cutoff you set will not be considered when constructing matrix. Default setting is 8
2) Minimal strand length: It is the length cutoff for selecting strand in structure, the strand with length < the cutoff you set will not be considered when constructing matrix. Default setting is 5
3) Interaction threshold: It is the distance cutoff for defining the interaction between one pair SSE. For one pair SSE except both of them are beta strands with H-bonding interaction (the interaction type will be c or t), the presence or absence of interaction is mainly defined by the distance, overlap and sheet information. The distance is the shortest distance between the C alpha coordinates on the two elements. SSEs with the interaction below the distance cutoff will be considered as have interaction, above that will be considered as "X" which means we do not care about if there is interaction or not. Default setting is 5 A. Setting it as small value, usually leads to more loose query matrix.
4) Minor strand: Default setting is do not consider minor strand when building query matrix.
After submitting the protein structure, an interactive page will appear. At this step, you can submit the query matrix directly, but it is often better to refine the query matrix to emphasize on the structure feature of the target motif you want to search for.
We provide three different option for user to refine the query matrix.
1) You can delete 'unimportant' SSEs by inputting SSE Index such as 1,2,3. You can view current interaction matrix to see what SSEs are in query matrix now with SSE range, SSE type, interaction type between SSE pair presented.
2) You can copy and paste the sheet info in text window and edit it by your self (sheet info can be get by 'view current sheet'). Submitting the sheet info will overwrite the old sheet info. If you want to delete all sheet info, just put 'nosheet' in the text window.
3) You can copy and paste the handedness info in text window and edit it by your self (sheet info can be get by 'view current handedness'). We enumerated all the handedness info for each consecutive triple SSE of your query structure. Submitting the handedness info will overwrite the old handedness info. If you want to delete all handedness info, just put 'nohandedness' in the text window.
In case that you do something wrong, 'Undo' operation is available for you to recover previous query matrix.
At the end of that interactive page, you can view current query matrix to see whole query matrix that you get. You can copy and paste it to the text window in "pattern search by matrix" (you can put more constraint there, for example, length requirement) and submit it or you can use 'submit current query matrix' to search against the structure database you selected.
"Check current query matrix" may be used for you to check the correctness of the query matrix you designed in case there is some obvious contradiction in your design.
Being a structure pattern search program, ProSMoS is not aimed at detecting homologs by structure similarity. All the found hits contain the user-defined structure pattern as exact matches, while all other proteins do not contain submatrix matching the query. Thus the results of ProSMoS are deterministic and binary: either a structure contains the pattern or it does not. However, ranking of hits is always beneficial for the users in order to focus on the most relevant results. Therefore, we developed a new but simple score function, which is used by the server to rank found hits. Our structure analysis experience indicates that motifs perceived as "good" by experts are compact, with regular and closely interacting SSEs, and SSE lengths are being close to their average values found in proteins. Hence, the scoring function is based on the distance and overlap between SSE pairs and SSE lengths, and is composed of three components: distance, overlap and length scores.
1) Distance score (D). First, we plotted the distance between SSE pairs for all proteins of PDB database and found that the average closest distance between adjacent β-strands is 4.5Å for parallel β-strands and 4.2Å for antiparallel β-strands, and the reasonable distance between neighboring β-strand-helix or helix-helix pair is 7.3Å for parallel SSEs and 7Å for antiparallel SSEs. Those values are set as the "ideal" distance between SSE pair. Second, we calculate the distance of each pair of SSE in motif hits if corresponding interaction has been specified in query matrix. Third, we sum over the squares of the differences between the observed distances and the ideal distances. The score is divided by the number of specified interactions plus one, and its square root is taken.
2). Overlap score (V). Considering the overlap definition described in the matrix construction section above, perfect overlap is equal to the vector length of the smaller SSE in a pair. First, we calculate the overlap for each SSE pair if interaction between them is defined in the query matrix. Then, we sum over the squares of the differences between overlap lengths and vector lengths of smaller SSEs. The score is also divided by the number of specified interactions plus one, and its square root is taken.
3) Length score (L). We define the length of SSE as the length of SSE vector in Å, rather than the number of residues of SSE. This makes all the three scores (D, V, and L) comparable and measured in Å. First, we computed all the lengths for β-strands and helices for the motif hits. Then, we calculate the median of β-strand length and the median of helix length. The median of SSE length represents the length tendency of SSE of the query structure pattern. Second, we sum over the squares of differences between observed SSE lengths and their median lengths. The score is divided by the number of SSEs in the query matrix plus one, and its square root is taken.
We combine the three scores in following function:
Score=1/[(WD* D + WV* V + L + 1].
Here, the weights WD, and Wv were set as 4.5 and 2.5 respectively to balance the three scores giving the best performance in our test. The weight of L is set to 1.0. Apparently, when all the sums of deviations from ideal values are close to 0, the score is close to 1 (maximum score). When the distances deviate from ideal, the score decreases to 0.
We display the result summary including the query matrix used, the target database selected, the number of pdb hits, the number of SCOP folds/superfamilies mapped etc. on the top of the main result. Then we comprehensively list every structure motif hits matching the query structure pattern ranking by the score as described in ranking score section. For each motif, the corresponding pdbid, protein name, SCOP domain id, description of domain, description of corresponding fold and superfamily and the motif SSE array are provided. The SSEs array of motif shows the information of pdb chain and SSEs pdb numbering range. Each hit can be visualized on the fly with PyMOL script which may help the user inspecting hits. The loaded PyMOL script displays the Cα trace of structure with the backbone colored in gray, the N-terminal start of motif colored in green, each secondary structure element colored in red and the first residue of each secondary structure element colored in purple.
Want to see the example output? Please click here
Any comments, suggestions or questions, please contact: shuoyong.shi@UTsouthwestern.edu