Database construction
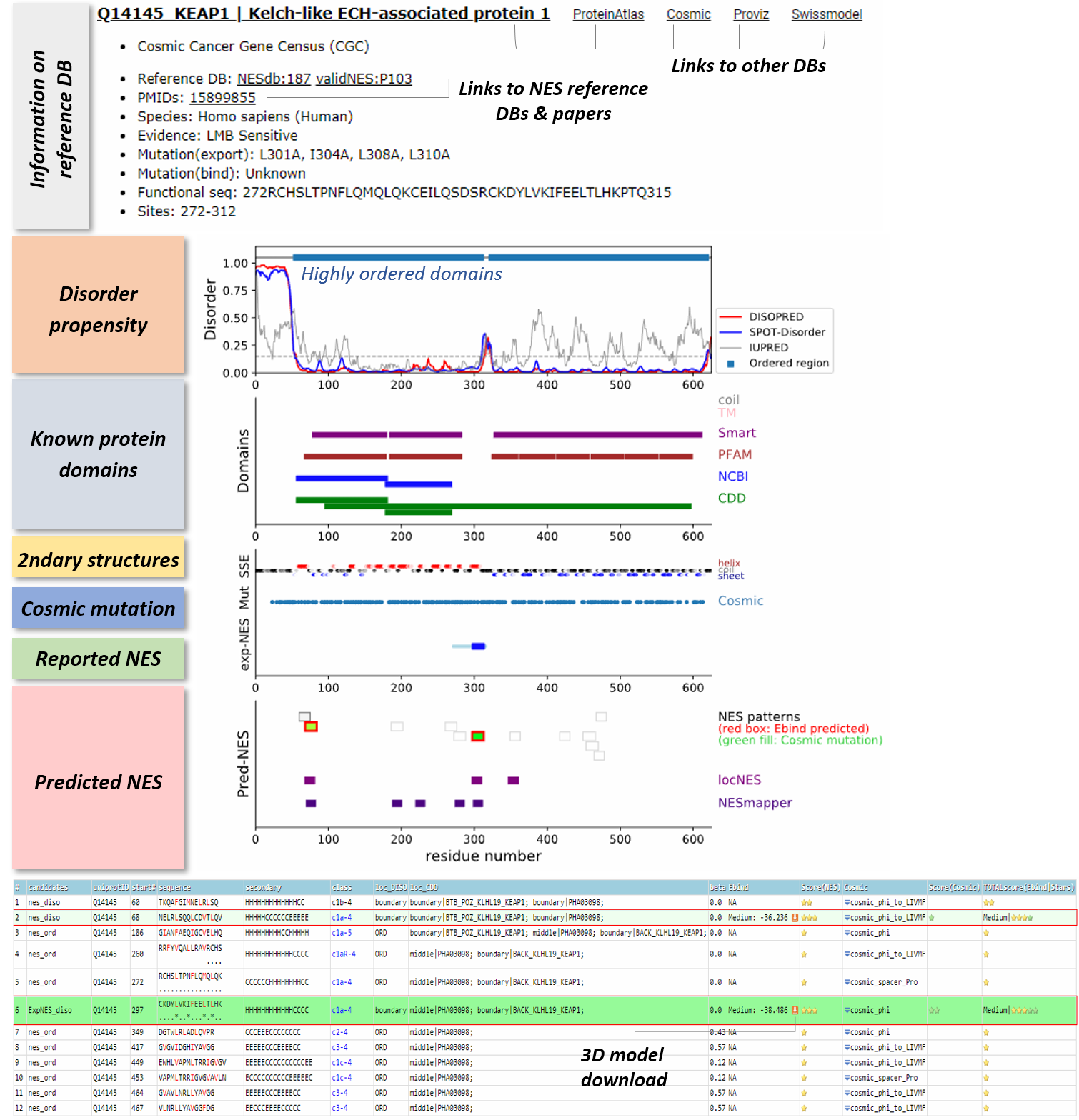
CRM1-dependent nuclear export signals (NESs), which regulate the active transport between the nucleus and cytoplasm for many cellular proteins, are short peptide sequences constituting of hydrophobic and spacer residues with specific patterns. We applied the combined sequence-based and structure-based approaches to analyze the NES motif regions' accessibility, conformation, and the stability at the binding site to the cancer-related human proteome sequences and provide a comprehensive database of the location of all NES consensus patterns, plotted together with the disorder propensity, known domain information, predicted secondary structures, binding scores of the segments in the CRM1's NES-binding groove, and the cancer-related mutation positions.
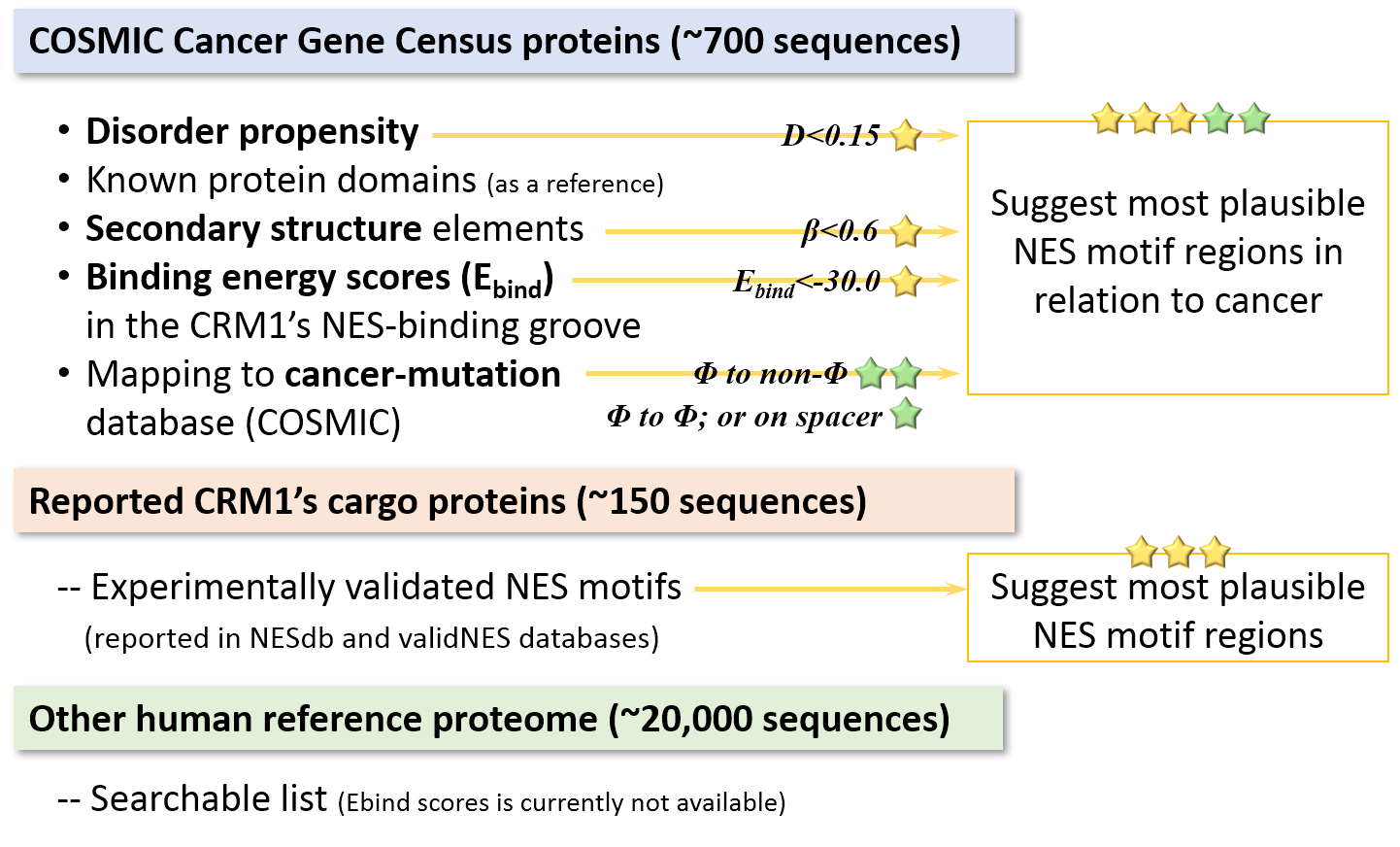
Scoring - STAR system
Each segment was scored via the STAR-system, i.e., how many criteria can be passed: the segments should i) not located in the ordered region, ii) not have beta strand in the middle, iii) have low or medium Ebind scores, and iv) have the hydrophobic positions in NES consensus overlapped with the cancer-related mutations.
Calculation details
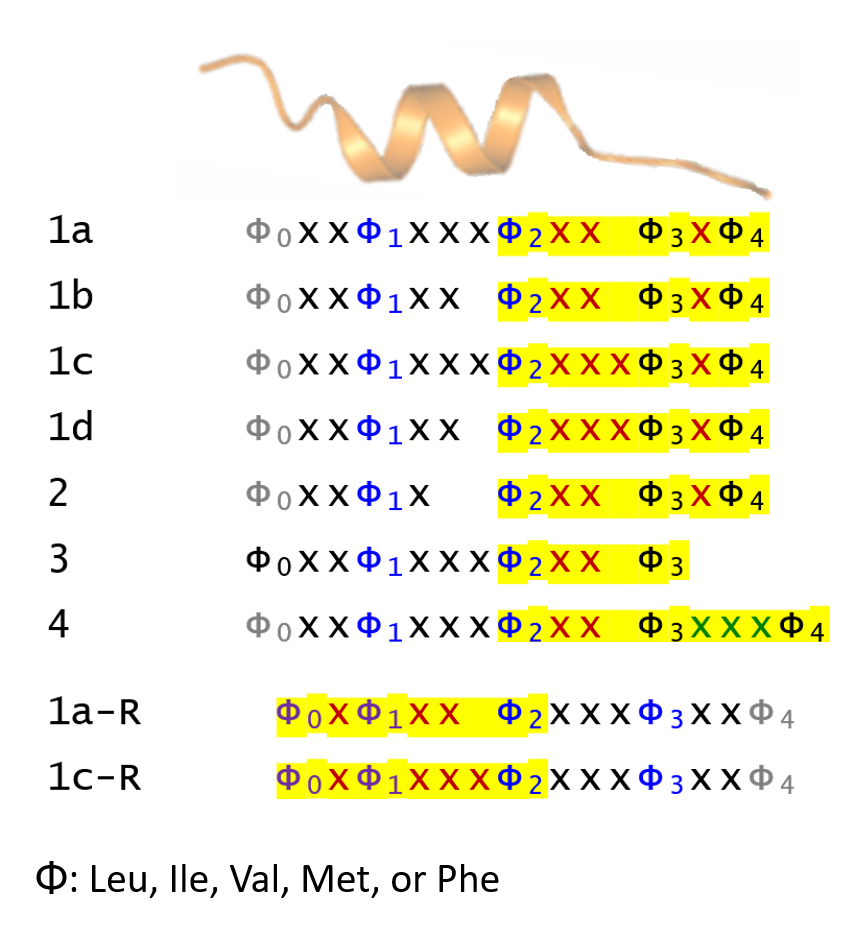
Extraction of the NES consensus sequences: Homo sapiens reference protein sequences were retrieved from Uniprot (Proteome ID: UP000005640). For NES consensus patterns, we utilized the modified version of the Kosugi consensus (Xu, et al., 2012; Xu, et al., 2015) and also refer the empirical class priority (for example, class 1a is the most abundant class; see (Lee, et al., 2019) for more details). If the Φ2-Φ4 portion of the extracted region overlaps with experimental evidence in NESdb or validNES, it is annotated as a candidate NES.
Disorder propensity: The disorder propensity of the cargo protein sequences is calculated using three different programs, DISOPRED3 (Jones and Cozzetto, 2015), SPOT-disorder (Hanson, et al., 2017), and IUPred2A (Meszaros, et al., 2018). Truly ordered and buried regions with high confidence are defined using the strict cutoff value (If a residue's disorder propensities predicted by both DISOPRED and SPOT-disorder are below 0.1, the residue is defined as ordered ("O"); if not, the residue is recorded as ("D"). If the portion of "D" mark is more than 90% for the segment and flanking regions (± 20 residues), the location of the segment (loc_DISO) is defined as an ordered region ("ORD"). If "D" is more than 90%, the location is determined as a disordered region ("DISO"). The other segments are considered as the ones located in the "boundary" region.
Known domains: By using Batch CD-search tool (Marchler-Bauer and Bryant, 2004), the conserved domain information for the cargo protein sequences was extracted. Four different databases, i.e., CDD (cdd v3.16), NCBI_Curated (cdd_ncbi v3.16), Pfam (oasis_pfam v3.16), SMART (oasis_smart v3.16), were searched with the expect value threshold of 0.01. The results were retrieved by the Concise mode. Transmembrane domains and coiled-coil regions are extracted by using UniProt annotation and marked together.
Secondary structure elements: Secondary structures of the cargo protein sequences are predicted by PSIPRED Version 4.02 (Jones, 1999). During PSI-BLAST search (Altschul, et al., 1997) to find homologs, uniref90_2015_01 (Suzek, et al., 2015) database is used. The confidence level of the prediction is also colored by a gradient from dark (high confidence) to light (low confidence).
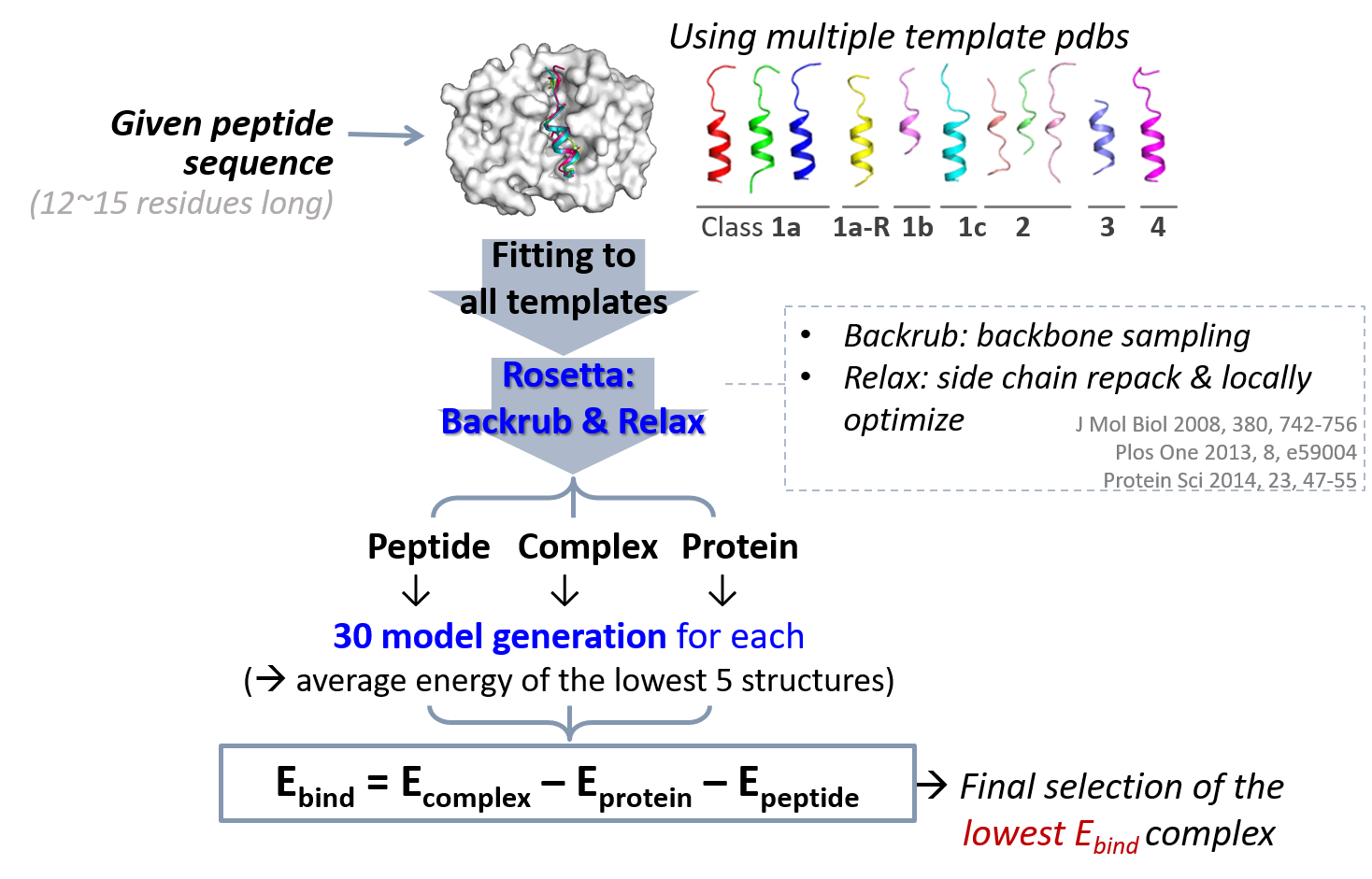
Relative binding energy (Ebind): A given peptide sequence is fitted to the backbone coordinates of every template structure. By using Rosetta Backrub (Smith and Kortemme, 2008) and Relax (Conway, et al., 2014; Nivon, et al., 2013) modules, the complex, the protein itself, and the free peptide structures are modeled separately with the same process. The binding energy (Ebind) is calculated by Ecomplex &minus Eprotein &minus Epeptide. For calculating Epeptide, we utilized the lowest energy among the all different backbone fitted models. Among the various template-fitted models, the one with the lowest Ebind score is finally selected.
Cancer-related mutation mapping: COSMIC mutation and Cancer Gene Census data was retrieved from https://cancer.sanger.ac.uk/cosmic (release v89). The meaningful mutation positions were selected using the following criteria: i) FATHMM prediction is 'pathogenic'; ii) Mutation somatic status is 'Confirmed somatic variant'; iii) Mutation description does not contain 'coding silent', 'frameshift', or 'nonsense'; and iv) the given position is not annotated as SNP.
References
[1] Lee, Y., et al. (2019) Structural prerequisites for CRM1-dependent nuclear export signaling peptides: accessibility, adapting conformation, and the stability at the binding site, Sci Rep, 9, 6627.
[2] Xu, D.R., et al. (2015) LocNES: a computational tool for locating classical NESs in CRM1 cargo proteins, Bioinformatics, 31, 1357-1365.
[3] Xu, D.R., Grishin, N.V. and Chook, Y.M. (2012) NESdb: a database of NES-containing CRM1 cargoes, Mol Biol Cell, 23, 3673-3676.
[4] Fu, S.C., et al. (2013) ValidNESs: a database of validated leucine-rich nuclear export signals, Nucleic Acids Res, 41, D338-D343.
[5] Conway, P., et al. (2014) Relaxation of backbone bond geometry improves protein energy landscape modeling, Protein Sci, 23, 47-55.
[6] Hanson, J., et al. (2017) Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks, Bioinformatics, 33, 685-692.
[7] Nivon, L.G., Moretti, R. and Baker, D. (2013) A Pareto-Optimal Refinement Method for Protein Design Scaffolds, Plos One, 8, e59004.
[8] Smith, C.A. and Kortemme, T. (2008) Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction, J Mol Biol, 380, 742-756.
[9] Jones, D.T. and Cozzetto, D. (2015) DISOPRED3: precise disordered region predictions with annotated protein-binding activity, Bioinformatics, 31, 857-863.
[10] Meszaros, B., Erdos, G. and Dosztanyi, Z. (2018) IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding, Nucleic Acids Res, 46, W329-W337.
[11] Marchler-Bauer, A. and Bryant, S.H. (2004) CD-Search: protein domain annotations on the fly, Nucleic Acids Res, 32, W327-W331.
[12] Jones, D.T. (1999) Protein secondary structure prediction based on position-specific scoring matrices, J Mol Biol, 292, 195-202.
[13] Suzek, B.E., et al. (2015) UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches, Bioinformatics, 31, 926-932.
[14] Altschul, S.F., et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res, 25, 3389-3402.
[15] Sondka, Z., et al. (2018) The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers, Nat Rev Cancer, 18, 696-705.
[16] Tate, J.G., et al. (2019) COSMIC: the Catalogue Of Somatic Mutations In Cancer, Nucleic Acids Res, 47, D941-D947.